本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- 人类癌症中致癌基因对非整倍性的成瘾
- 开发用于突触核蛋白病成像的 α-突触核蛋白正电子发…
- TMEM106B 是介导不依赖 ACE2 的 SA…
- 染色质扩展显微镜研究纳米级核组织
- 以高分辨率可视化的人类细胞翻译动力学揭示癌症药物作…
前言
本文是前沿快讯的第33期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。从第18期开始,“肿瘤转移类”、“肿瘤代谢类”等将不再更新,而是合并至其它分类。
本期有以下知识点值得关注:
CNS类
人类癌症中致癌基因对非整倍性的成瘾
Oncogene-like addiction to aneuploidy in human cancers. Science
我一直想知道答案的问题。十分有趣。
- 大多数癌症表现出非整倍性,但其在肿瘤发展中的功能意义存在争议。
- 在这里,我们描述了 ReDACT(使用 CRISPR 靶向恢复非整倍体细胞中的二倍体),这是一组染色体工程工具,使我们能够消除癌症基因组中的特定非整倍体。
- 使用 ReDACT,我们创建了一组具有或缺乏常见非整倍性的同基因细胞,并且我们证明 1q 染色体三体性对于具有这种改变的癌症的恶性生长是必需的。从机制上讲,获得染色体 1q 会增加 MDM4 的表达并抑制 p53 信号传导,我们表明 TP53 突变与人类癌症中的 1q 非整倍体是相互排斥的。
- 因此,肿瘤细胞可能依赖于特定的非整倍性,从而提高了将这些“非整倍性成瘾”作为治疗策略的可能性。
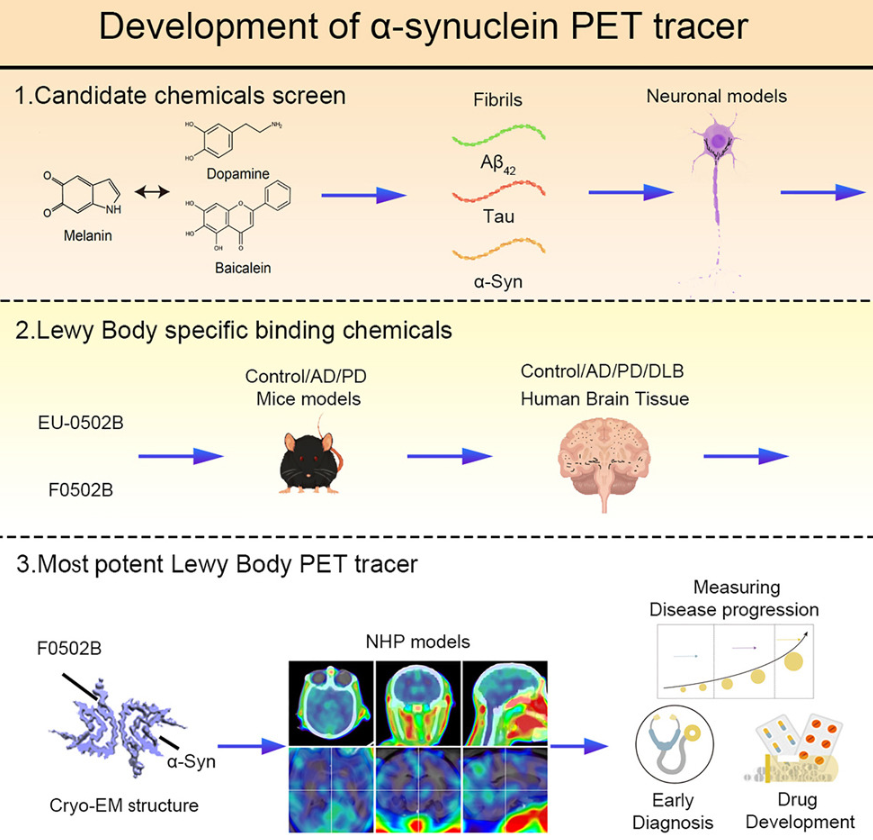
开发用于突触核蛋白病成像的 α-突触核蛋白正电子发射断层扫描示踪剂
Development of an α-synuclein positron emission tomography tracer for imaging synucleinopathies. Cell
- 突触核蛋白病的特征是 α-突触核蛋白 (α-Syn) 在大脑中聚集。突触核蛋白病的正电子发射断层扫描 (PET) 成像需要选择性结合 α-Syn 沉积物的放射性药物。
- 我们报告了一种脑可渗透且快速冲洗的 PET 示踪剂 [18F]-F0502B 的鉴定,该示踪剂对 α-Syn 显示出高结合亲和力,但对 Aβ 或 Tau 纤维没有,并且优先与脑切片中的 α-Syn 聚集体结合。通过对来自多个小鼠模型和人类受试者的体外原纤维、神经元内聚集体和神经退行性疾病脑切片进行多个周期的反筛选,[18F]-F0502B 对小鼠和非人类灵长类 PD 模型大脑中的 α-Syn 沉积物进行成像。
- 我们进一步通过冷冻电镜确定了 α-Syn 原纤维-F0502B 复合物的原子结构,并通过配体间相互作用通过强烈的非共价键合网络揭示了 F0502B 在原纤维表面上的平行对角堆积。
- 因此,[18F]-F0502B 是一种有前途的先导化合物,用于对突触核蛋白病中聚集的 α-Syn 进行成像。
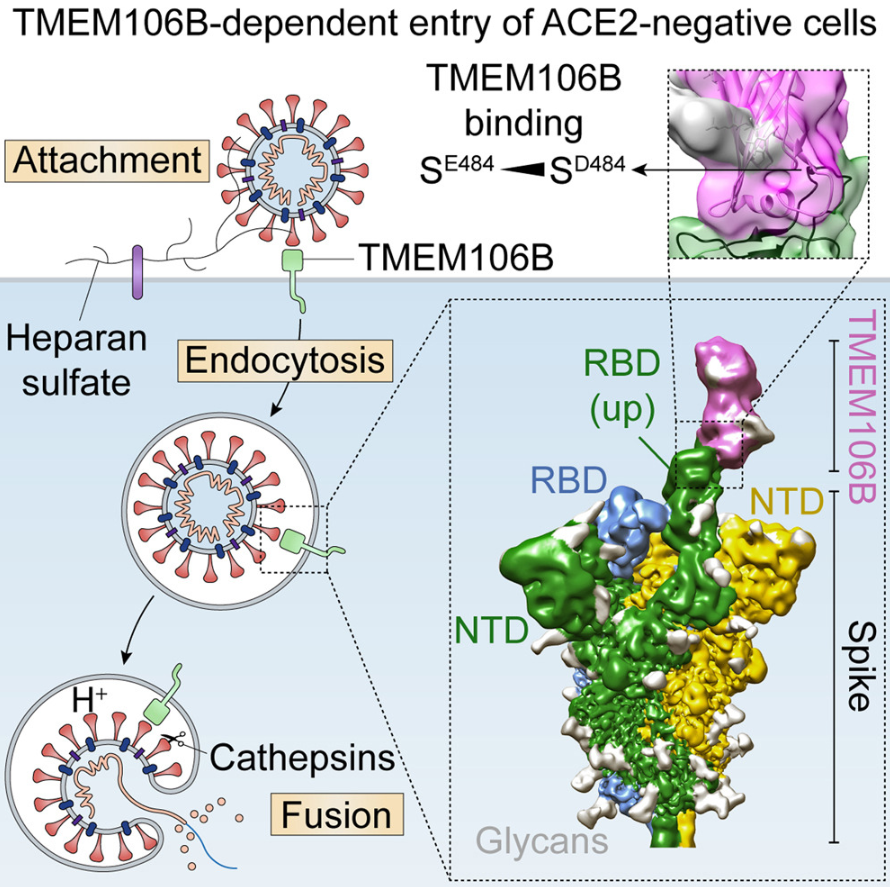
TMEM106B 是介导不依赖 ACE2 的 SARS-CoV-2 细胞进入的受体
TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell
- SARS-CoV-2 与广泛的组织趋向性相关,这一特征通常由宿主细胞上进入受体的可用性决定。
- 在这里,我们证明 TMEM106B(一种溶酶体跨膜蛋白)可以作为 SARS-CoV-2 进入血管紧张素转换酶 2 (ACE2) 阴性细胞的替代受体。刺突取代 E484D 增加 TMEM106B 结合,从而增强 TMEM106B 介导的进入。 TMEM106B 特异性单克隆抗体可阻断 SARS-CoV-2 感染,证明 TMEM106B 在病毒进入中的作用。
- 使用 X 射线晶体学、低温电子显微镜 (cryo-EM) 和氢氘交换质谱 (HDX-MS),我们表明 TMEM106B 的管腔结构域 (LD) 与 SARS-CoV-的受体结合基序接合。最后,我们证明 TMEM106B 促进刺突介导的合胞体形成,表明 TMEM106B 在病毒融合中的作用。
- 总之,我们的研究结果确定了一种不依赖于 ACE2 的 SARS-CoV-2 感染机制,该机制涉及与受体硫酸乙酰肝素和 TMEM106B 的协同相互作用。
染色质扩展显微镜研究纳米级核组织
Chromatin expansion microscopy reveals nanoscale organization of transcription and chromatin. Science
- 纳米级染色质组织调节基因表达。尽管染色质在合子基因组激活(zygotic genome activation, ZGA)过程中发生了显着的重新编程,但在这一普遍过程中染色质调节因子的组织仍不清楚。
- 在这项工作中,我们开发了染色质扩增显微镜 (chromatin expansion microscopy, ChromExM),用于可视化体内染色质、转录和转录因子。
- ZGA 期间胚胎的 ChromExM 揭示了先驱因子 Nanog 如何与核小体和 RNA 聚合酶 II (Pol II) 相互作用,提供了转录延伸作为串状纳米结构的直接可视化。阻断延伸导致更多的 Pol II 颗粒聚集在 Nanog 周围,Pol II 停滞在启动子和 Nanog 结合的增强子处。这导致了一种称为“kiss and kick”的新模型,其中增强子-启动子接触是短暂的,并通过转录延伸释放。
- 我们的结果表明 ChromExM 广泛适用于研究纳米级核组织。
以高分辨率可视化的人类细胞翻译动力学揭示癌症药物作用
Translation dynamics in human cells visualized at high resolution reveal cancer drug action. Science
本研究研究药物处理后核糖体结构的方法与直接解析蛋白质结构的方法有何不同?
- 核糖体通过循环各种功能状态来催化蛋白质合成。这些状态已在体外得到广泛表征,但它们在主动翻译的人类细胞中的分布仍然难以捉摸。
- 我们使用基于冷冻电子断层扫描的方法,以高分辨率解析了人体细胞内的核糖体结构。这些结构揭示了延伸循环的功能状态分布、Z 转移 RNA 结合位点以及核糖体扩展片段的动态。
- 用高三尖杉酯碱(一种用于治疗慢性粒细胞白血病的药物)处理的细胞的核糖体结构揭示了翻译动力学如何原位改变并解析核糖体活性位点内的小分子。
- 因此,可以在人体细胞内以高分辨率评估结构动力学和药物作用。
皮质极性确保气孔谱系中自身的不对称遗传从而形成叶子表面的图案
- 不对称细胞分裂指定了不同细胞界的不同细胞命运。在后生动物中,命运决定因素优先遗传到一个子细胞中通常取决于极性-细胞骨架相互作用。尽管在整个植物发育过程中普遍存在不对称分裂,但分离命运决定因素的类似机制的证据仍然难以捉摸。
- 在这里,我们描述了拟南芥叶表皮中的一种机制,该机制确保命运执行极性域的不平等遗传。通过定义一个缺乏稳定微管的皮质区域,极性域限制了可能的分裂方向。因此,有丝分裂期间将极性域与微管组织解偶联会导致异常的分裂平面和伴随的细胞身份缺陷。
- 我们的数据强调了如何重新配置一个常见的生物模块,通过细胞骨架将极性与命运分离耦合起来,以适应植物发育的独特特征。
人类pre-60S的生物发生原理
- 核糖体是两个亚基 RNA-蛋白质纳米机器,可将信使 RNA (mRNA) 翻译成所有生物体中的蛋白质。小核糖体亚基 (40S) 负责 mRNA 的解码,而大亚基 (60S) 催化肽键形成。人体细胞中两个核糖体亚基的组装都是在核仁中启动的,随后是核和细胞质的成熟,并且需要 200 多个核糖体组装因子来催化核糖体 RNA (rRNA) 的修饰、加工和折叠。在核仁组装过程中,大核糖体亚基前体(pre-60S)由 5S rRNA 和 32S pre-rRNA 前体组装而成,后者包含通过内部转录间隔区 2 (ITS2) 连接的 5.8S rRNA 和 28S rRNA。由于无法分离早期内源性 60S 前组装中间体,迄今为止对人类大核糖体亚基组装的了解仅限于非常晚期的核状态。因此,人类 60S 前粒子的核仁组装和核成熟的机制仍然未知。
- 我们着手从机械层面阐明 60S 前人类组装的早期阶段。为此,我们将人类基因组编辑和生物化学相结合,对人类核仁和细胞核进行透化,以分离完整的内源性 60S 前组装中间体,以便通过冷冻电子显微镜 (cryo-EM) 进行后续结构表征。为了对 rRNA 加工进行功能性研究,我们开发了一种体内重组核糖体组装测定法,使用工程化的人类 rDNA 基因座来研究 ITS2 和 28S rRNA 中的 rRNA 元件是否是大核糖体亚基生物发生所必需的。
- 在这项研究中,我们对 ITS2 相关组装因子 MK67I 进行双等位亲和标记,并以 2.5 至 3.2 Å 的分辨率确定了 60S 前人类组装中间体的 24 个冷冻电镜结构。在这种结构景观中,我们观察到了几种平行的组装途径,并确定了八种主要的核仁组装状态和四种主要的核成熟状态。在核仁中,这些结构突出了蛋白质相互作用中心如何将组装因子复合物连接到成熟的 60S 前颗粒,以及 GTP 酶和 ATP 酶(例如 DEAD-box RNA 解旋酶)如何将不可逆的核苷酸水解步骤与功能中心的安装相结合。离开核仁后,成熟的 pre-60S 颗粒展示了 pre-rRNA 的结构可塑性如何受到包括核糖体在内的一组组装因子的影响,以将 rRNA 构象变化与 RNA 外泌体介导的 ITS2 降解结合起来。后来的 60S 前核状态揭示了如何使用组装因子交换来驱动功能中心的不可逆微调。此外,通过使用工程化的人类 rRNA,我们已经确定了 ITS2 和 28S rRNA 的元件,这些元件对于在人类细胞中生成成熟的大核糖体亚基至关重要。我们的冷冻电镜重建的高分辨率进一步使我们能够可视化许多先前绘制的人类 28S rRNA 化学修饰,为研究普遍保守的化学修饰以及作为核糖体组装功能的物种特异性适应奠定了基础。
- 我们的冷冻电镜结构整体与我们工程化的核糖体组装检测一起,现在为人类大核糖体亚基的核仁组装和核成熟提供了高分辨率的视角。我们的研究合理化了数十年的遗传和生化数据,并揭示了真核核糖体组装的新原理。
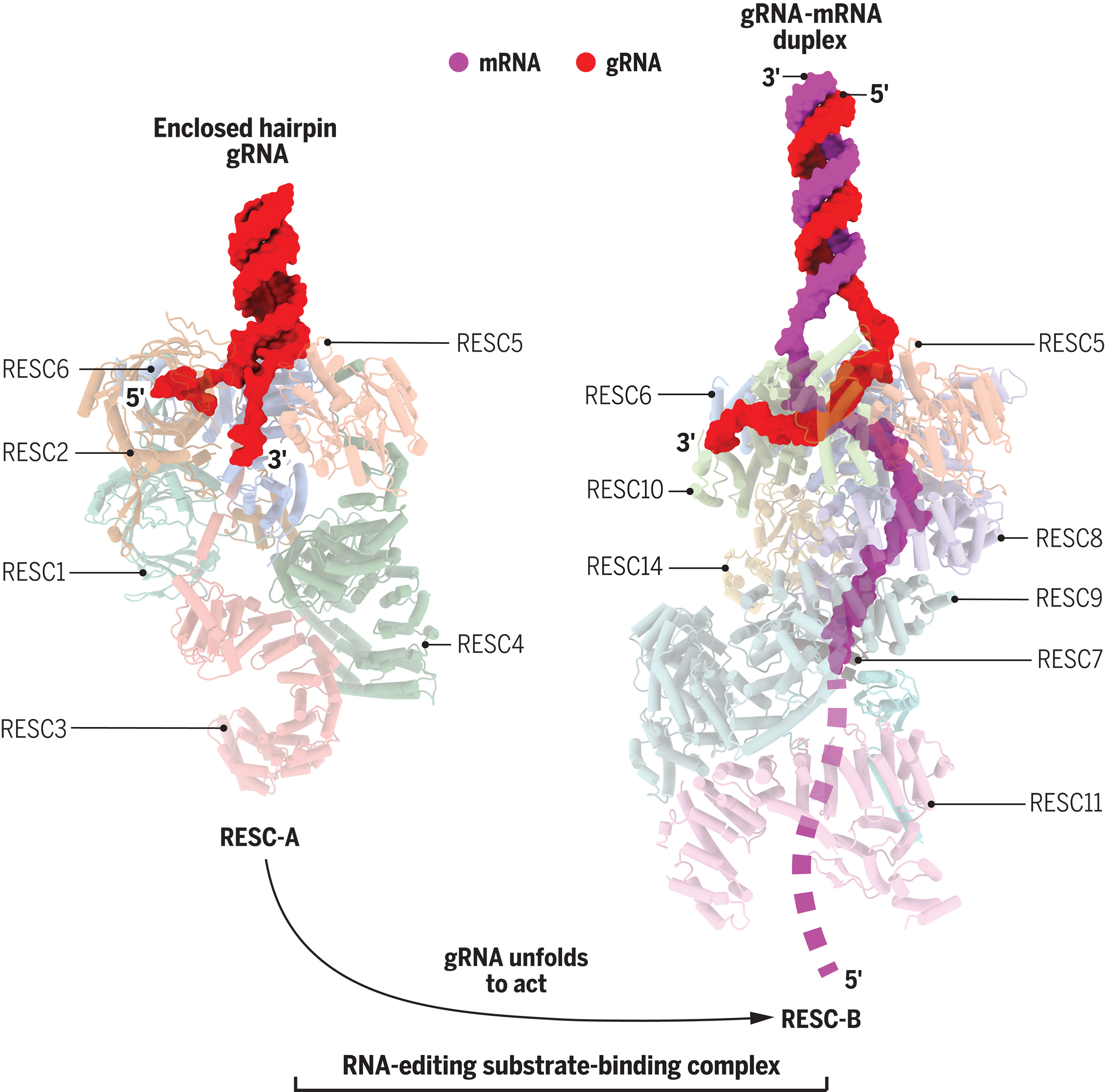
锥虫 RNA 编辑中 gRNA 稳定和 mRNA 识别的结构基础
Structural basis of gRNA stabilization and mRNA recognition in trypanosomal RNA editing. Science
- RNA 编辑是在布氏锥虫(一种导致非洲昏睡病的原生动物寄生虫)的线粒体中发现的,它表示一系列系统发育上广泛存在且通常在机制上不相关的改变 RNA 序列的分子过程。在锥虫 RNA 编辑中,gRNA 指导大量重新编码神秘的线粒体转录物以生成 mRNA。两个主要核糖核蛋白颗粒组装到部分定义的编辑体中,执行从 gRNA 到 mRNA 遗传信息转移的非常规方法。 RNA 编辑底物结合复合物 (RESC) 可稳定 gRNA 并与 mRNA 结合。 RNA 编辑催化复合物 (RECC) 完成 gRNA 编程的 mRNA 切割、尿苷插入或缺失以及重新连接反应。
- 线粒体 gRNA 与 mRNA 前体形成不完美的双链体,其中二级结构定义了多个编辑位点。为了揭示 gRNA 稳定性和 mRNA 识别机制,我们使用低温电子显微镜确定了 RESC 三种状态的原子结构,并表征了各个亚基的 RNA 结合特异性。
- 生化研究将 RESC 定义为包含 gRNA 和 mRNA 的约 18 种蛋白质的异质组装体。先前的工作还表明,RESC1/2 二聚体可以稳定 gRNA 和大多数其他蛋白质结合 mRNA。六元RESC-A的结构显示了RESC2 RNA三磷酸酶假酶如何吞噬三磷酸化gRNA的5’端,以及RESC5/6二聚体如何固定3’端。这些接触促进 gRNA 折叠成“发夹样”构象,并保护两个末端免受核酸酶的影响。 10-多肽RESC-B结构表明重塑事件从RESC-A“存储”模式恢复gRNA并将单链分子转变为mRNA邻近状态。在此过程中,gRNA 的 5′ 末端从 RESC2 三磷酸结合通道中弹出,但 3′ 末端仍楔入 RESC5 和 RESC6 之间,这是 RESC-A 和 RESC-B 之间唯一共享的蛋白质。所有RESC-B 亚基,包括RESC5/6,都沿着约20 个核苷酸的片段接触mRNA。然而,gRNA 和 mRNA 在 RESC-B 边界内不相互作用。
- 典型的 gRNA 以与 mRNA 靶点完全互补的“锚”开始;相邻的“引导”部分与 mRNA 稀疏配对,并创建以单链凸起和环为代表的编辑位点。一些核苷酸将这个“信息丰富”的序列与末端尿苷尾分开。从机制上讲,将 gRNA 的“信息贫乏”3′ 端隔离在 RESC-B 内部,可以引导和锚定部分与 RESC-B 表面之外的 mRNA 杂交。显然,RESC-B 蛋白会招募 gRNA 和 mRNA,无论其序列如何,并将两条链以大致反平行方向定位,但距离足以防止复合物内的虚假退火。暴露的 gRNA 和 mRNA 区域可能会互相采样,直到有效杂交为催化 RECC 复合物创造底物。
- 总之,不同 RESC 状态的结构揭示了已被纳入 RNA 编辑机制的蛋白质折叠的多样性。我们已经发现了常见的 gRNA 元件如何发挥稳定这种短分子和接合 mRNA 的作用。我们的结果表明,gRNA-mRNA 识别源自核糖核蛋白复合物重塑,而不是启动锚定序列与 mRNA 靶标的碱基配对。最后,有关基本功能特征的结构信息,例如 RESC2 三磷酸结合位点,可能有助于新锥虫杀剂的开发。
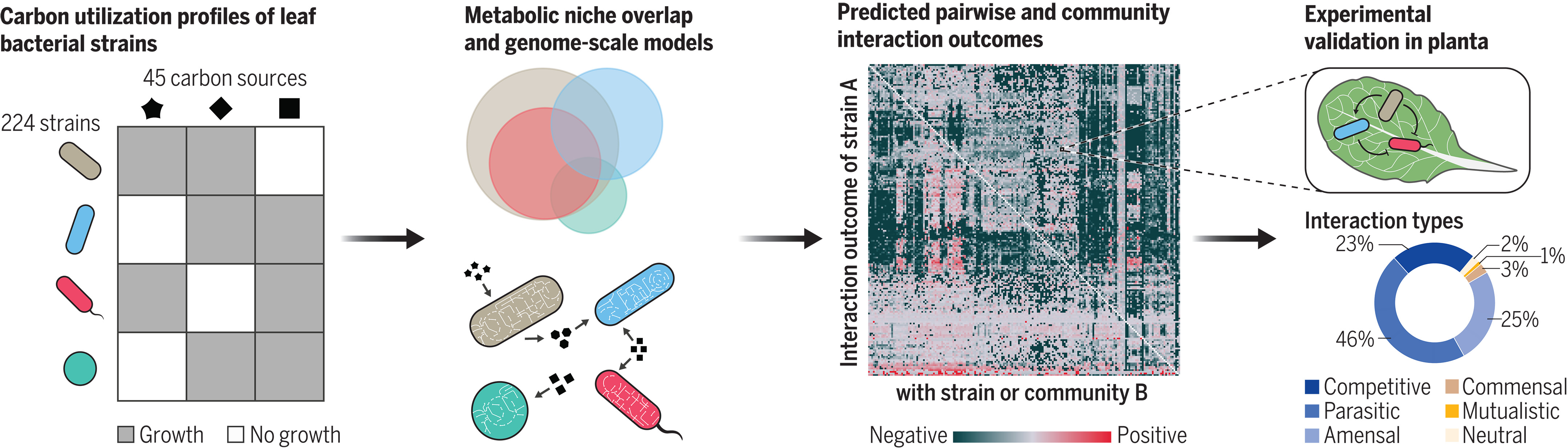
代谢相互作用模型概括了叶片微生物群生态学
Metabolic interaction models recapitulate leaf microbiota ecology. Science
- 微生物组的组成与宿主健康和生态系统功能有关。然而,人们对环境中决定物种丰度的过程仍然知之甚少。鉴于微生物组受到资源可用性的限制,对其组成生物体的代谢能力的深入了解为预测种间相互作用和更广泛的微生物组组装规则提供了关键途径。植物构成了地球上最大的生物量,并且被各种影响其健康和生长的微生物定殖。人们发现植物微生物组的组成在很大程度上是确定性的,这表明存在确定的群落组装驱动因素。然而,这些模式背后相互作用的代谢机制仍然知之甚少。
- 为了评估资源分配模式对微生物相互作用的影响程度,我们首先分析了从野生拟南芥植物叶子中分离出的 224 种代表性细菌菌株的碳源利用能力。为此,我们在 45 种不同的碳源上培养了每种菌株,并开发了一种计算工具,以自动方式对每种菌株的生长进行评分。该筛选使我们能够计算菌株之间代谢生态位重叠的程度,从而预测植物宿主的资源竞争。我们进一步利用这些实验数据生成了 224 个基因组规模代谢模型的集合,其中涵盖了每个生物体的代谢网络。我们使用这些模型来预测植物宿主上物种间相互作用的结果,并获得对这些细菌之间资源利用和交换的特定模式的机制洞察。
- 我们发现,基于实验筛选和基因组规模模型中代表的途径,菌株的碳源利用概况表现出强烈的系统发育特征。我们通过在拟南芥宿主上用两组菌株进行竞争实验,验证了模型的性能和基于生态位重叠的预测。这些实验揭示了负面相互作用结果的主导地位(即,与单一关联相比,菌株在共定殖时达到较低的总体种群水平),这与菌株间生态位高度重叠的预测一致。基因组规模模型提供了对这些相互作用的额外了解,还正确预测了在植物中观察到的积极结果的实例,并进一步强调了碳代谢对群落组装的重要性。经过实验验证后,我们对 17,500 多对菌株的相互作用结果进行了建模。我们预测,在 94% 的配对中,与单关联相比,至少有一个菌株的丰度会减少。然后,我们分析了这些预测结果背后的代谢通量,这揭示了糖竞争的高度普遍性。这种竞争可以通过我们的模拟中氨基酸和有机酸的吸收增加来抵消,这表明代谢机制支撑着我们在植物中观察到的积极相互作用结果。
- 我们的结果表明,碳源可用性和代谢相互作用在植物宿主的群落组装中发挥着重要作用,再加上此处观察到的性状保存,可能有助于植物微生物组组装的总体确定性结果。除了现场重现种间相互作用外,我们的建模框架还提供了一个强大的工具,用于确定导致特定生态模式出现的代谢机制。这些知识最终将实现有针对性的微生物组设计,这对于微生物组在健康、农业和环境中的应用至关重要。
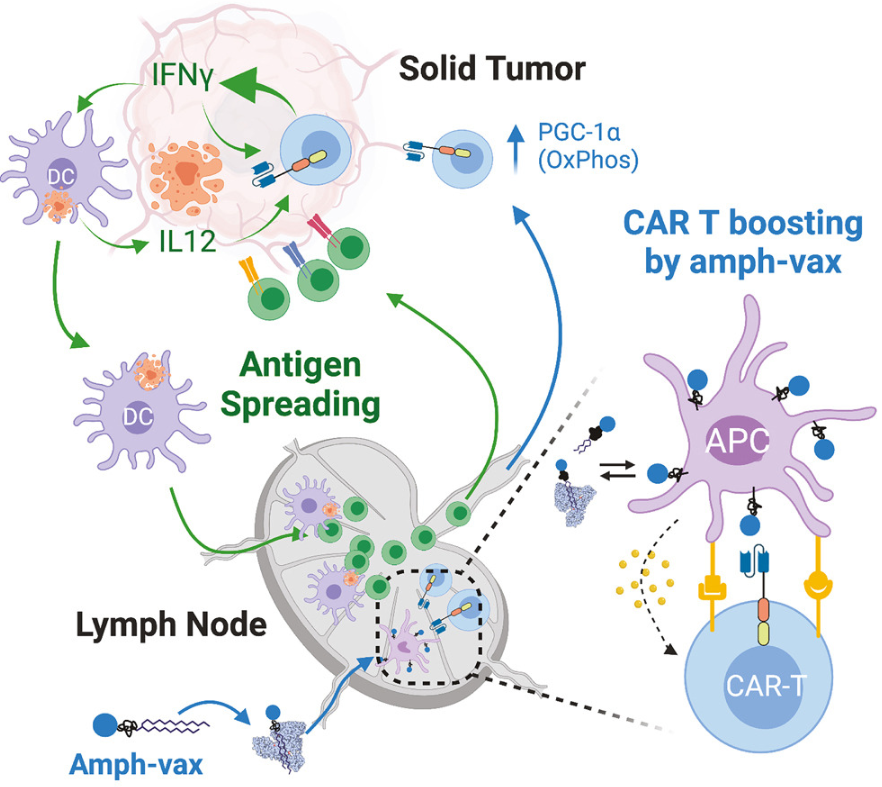
疫苗增强的 CAR-T 与宿主免疫串扰以排斥具有抗原异质性的肿瘤
Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell
听上去挺神奇的
- 嵌合抗原受体 (CAR) T 细胞疗法可有效治疗人类癌症,但 CAR 识别的抗原的丧失构成了主要障碍。
- 我们发现,体内 CAR T 细胞的疫苗加强会触发内源性免疫系统的参与,从而避免抗原阴性的肿瘤逃逸。疫苗增强的 CAR T 促进树突状细胞 (DC) 招募至肿瘤,增加 DC 对肿瘤抗原的摄取,并引发内源性抗肿瘤 T 细胞的启动。这一过程伴随着 CAR-T 代谢向氧化磷酸化 (OXPHOS) 的转变,并且严重依赖于 CAR-T 衍生的 IFN-γ。
- 即使初始肿瘤 50% CAR 抗原阴性,疫苗增强的 CAR T 诱导的抗原扩散 (AS) 也能实现一定比例的完全缓解,并且通过 CAR T IFN-γ 表达的基因扩增进一步增强异质性肿瘤控制。
- 因此,CAR-T 细胞衍生的 IFN-γ 在促进 AS 中发挥着关键作用,而疫苗加强提供了一种临床上可转化的策略来驱动针对实体瘤的这种反应。
人类胚胎实时成像揭示囊胚扩张和活检过程中核 DNA 脱落
Human embryo live imaging reveals nuclear DNA shedding during blastocyst expansion and biopsy. Cell
该实验观察的是人类子宫里的胚胎还是实验室里的胚胎?
- 适当的植入前发育对于组装能够植入的囊胚至关重要。实时成像揭示了驱动小鼠胚胎早期发育的重大事件;然而,由于基因操作的限制和缺乏成像方法,人类研究受到限制。
- 我们通过将荧光染料与实时成像相结合,克服了这一障碍,揭示了人类胚胎中染色体分离、压缩、极化、囊胚形成和孵化的动态。我们还表明囊胚扩张机械地限制滋养外胚层细胞,导致核出芽和 DNA 脱落到细胞质中。此外,核周角蛋白水平较低的细胞更容易发生 DNA 丢失。此外,应用滋养外胚层活检(一种临床上用于基因检测的机械程序)会增加 DNA 脱落。
- 因此,我们的工作揭示了与小鼠相比人类发育的不同过程,并表明人类胚胎中的非整倍体可能不仅源于有丝分裂期间的染色体分离错误,而且还源于核DNA脱落。
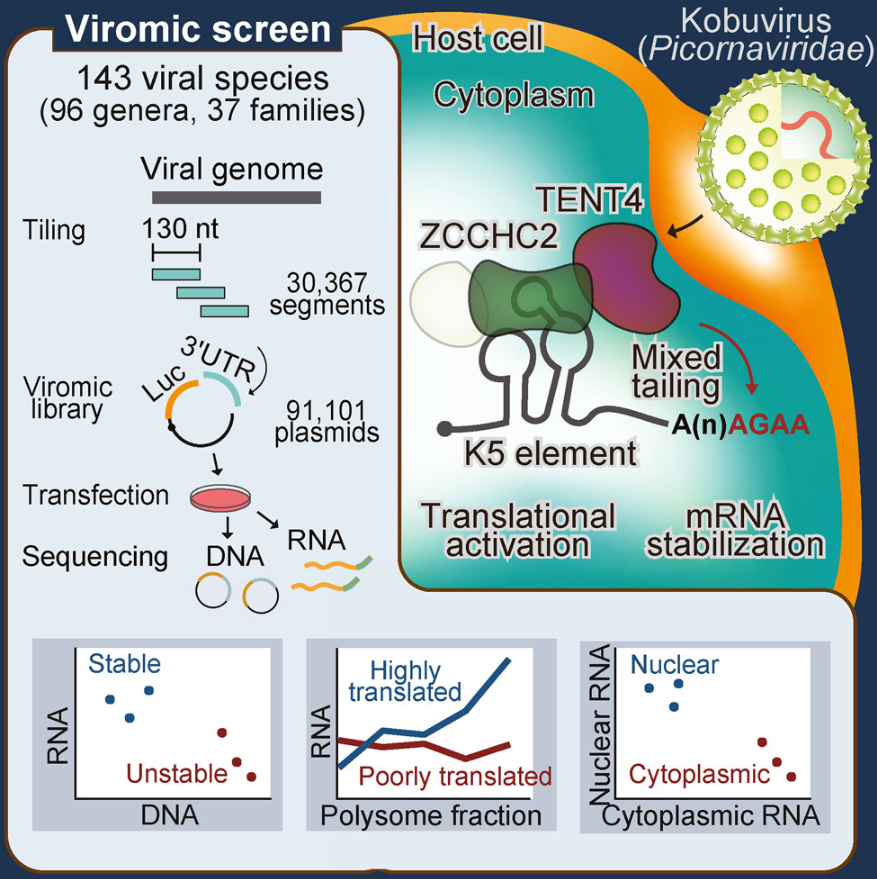
功能性病毒组筛选揭示调控 RNA 元件
Functional viromic screens uncover regulatory RNA elements. Cell
- 最近,已测序的病毒基因组数量激增,为了解病毒多样性和揭示未知的调控机制提供了机会。在这里,我们对来自 37 个科 96 个属 143 个物种的 30,367 个病毒片段进行了筛选。利用 3′ UTR 中的病毒片段文库,我们鉴定了数百个影响 RNA 丰度、翻译和核质分布的元件。
- 为了说明这种方法的威力,我们研究了 K5(一种在科布病毒中保守的元件),发现它在各种情况下(包括腺相关病毒载体和合成 mRNA)增强 mRNA 稳定性和翻译的强大能力。此外,我们还发现了一种以前未表征的蛋白质 ZCCHC2,它是 K5 的关键宿主因子。 ZCCHC2 招募末端核苷酸转移酶 TENT4 以延长具有混合序列的聚腺苷酸尾,从而延迟去腺苷化。
- 这项研究为病毒和 RNA 研究提供了独特的资源,并强调了病毒圈在生物发现方面的潜力。
oviDN神经元调节果蝇产卵决策的机制
A rise-to-threshold process for a relative-value decision. Nature
- 尽管在识别与快速提示决策相关的神经信号方面已经取得了进展,但人们对大脑如何引导和终止与动物行为学相关的更多决策知之甚少,在这些决策中,动物自身的行为控制着在几分钟内经历的选择。果蝇需要花费数秒到数分钟的时间来寻找具有较高相对价值的产卵位点,并且具有称为oviDN的神经元,其活动满足启动产卵运动程序的必要性和充分性标准。
- 在这里,我们表明,oviDN 表达钙信号,该信号(1)在卵子内部准备(排卵)时下降,(2)在几秒到几分钟内上下漂移(其方式受底物相对值的影响),就像果蝇一样确定是否产卵,并且 (3) 在腹部弯曲产卵之前达到一致的峰值水平。该信号在大脑中 oviDN 的细胞体中很明显,它可能反映了腹神经索中与行为相关的上升至阈值过程,oviDN 的突触末端位于此处,其输出可以影响行为。
- 我们提供的扰动证据表明,一旦该过程达到阈值,卵沉积运动程序就会启动,并且该过程中的阈下变化调节考虑选项所花费的时间以及最终所采取的选择。最后,我们确定了一个小的循环回路,该回路可馈入 oviDN,并表明其每种组成细胞类型的活性都是产卵所必需的。
- 这些结果表明,上升到阈值的过程调节相对价值、自定进度的决策,并为构建该过程的底层电路机制提供了初步见解。
最小细胞的进化之路
Evolution of a minimal cell. Nature
- 最小的细胞只拥有必需的基因,可以揭示对生命的持久性和稳定性至关重要的机制和过程。在这里,我们报告了与合成衍生的丝状支原体非最小细胞相比,工程化最小细胞如何对抗进化的力量。
- 突变率在所有报道的细菌中最高,但不受基因组最小化的影响。基因组精简成本高昂,导致适应性下降超过 50%,但这种缺陷在 2000 代进化过程中又得到了恢复。尽管选择作用于不同的遗传目标,但合成细胞的最大生长速率的增加是相当的。此外,当通过相对适应性评估表现时,最小细胞的进化速度比非最小细胞快 39%。唯一明显的限制涉及细胞尺寸的演变。非最小细胞的大小增加了 80%,而最小细胞保持不变。这种模式反映了 ftsZ 突变的上位效应,ftsZ 编码调节细胞分裂和形态的微管蛋白同源蛋白。
- 我们的研究结果表明,自然选择可以迅速提高最简单的自主生长生物体之一的适应性。了解具有小基因组的物种如何克服进化挑战,为宿主相关内共生体的持久性、生物技术流线型底盘的稳定性以及合成工程细胞的有针对性的细化提供了重要的见解。
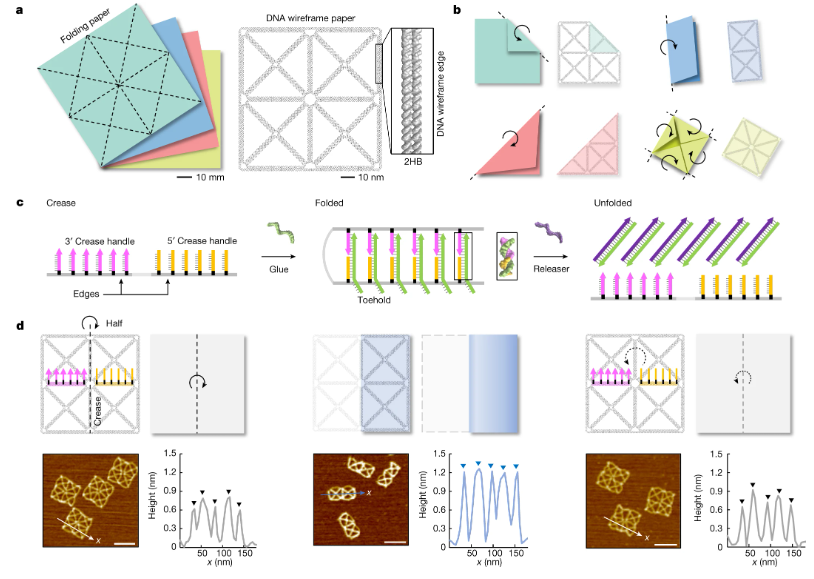
利用纸张折叠机制进行可重构 DNA 折纸
Harnessing a paper-folding mechanism for reconfigurable DNA origami. Nature
韩国首尔国立大学-机械工程系- 先进机械与设计研究所
比较少见但很有趣的研究
- 折纸机构因其在将可变形状和刚度编程到结构中的独特能力和优势而被广泛应用于构建可重构宏观系统。然而,由于缺乏合适的设计原理,尽管已经开发了各种基于 DNA 自组装的动态结构,但由于缺乏合适的设计原理,它几乎没有被用于分子级系统的构建。
- 在这里,我们提出了一种利用纸张折叠机制来创建可重构 DNA 折纸结构的方法。主要思想是构建一个参考平面线框结构,其边缘遵循纸张折叠中的折痕图案,以便可以折叠成各种目标形状。我们利用 DNA 链置换实现了几种类似纸张的折叠和展开模式,并且产量很高。演示了正交折叠、可重复折叠和展开、基于折叠的 microRNA 检测和荧光信号控制。由 pH 值或光源变化触发的刺激响应折叠和展开也是可能的。此外,通过采用分层组装,我们可以以高度可编程的方式扩展折纸机构的设计空间和复杂性。
- 由于其高度的可编程性和可扩展性,我们期望所提出的基于纸张折叠的重构方法将促进复杂分子系统的发展。
cDC1 中的 SLC38A2 和谷氨酰胺信号传导决定抗肿瘤免疫
SLC38A2 and glutamine signalling in cDC1s dictate anti-tumour immunity. Nature
- 癌细胞通过肿瘤-免疫相互作用逃避 T 细胞介导的杀伤,但其机制尚不清楚。树突状细胞 (DC),尤其是 1 型常规 DC (cDC1),介导 T 细胞启动和针对肿瘤的治疗功效。 DC 功能由模式识别受体协调,但涉及的其他信号仍未完全确定。营养素是适应性免疫的新兴介质,但营养素是否影响 DC 功能或先天性免疫细胞和适应性免疫细胞之间的通讯在很大程度上尚未解决。
- 在这里,我们将谷氨酰胺建立为细胞间代谢检查点,决定肿瘤-cDC1 串扰并许可 cDC1 激活细胞毒性 T 细胞的功能。瘤内补充谷氨酰胺通过增强 cDC1 介导的 CD8+ T 细胞免疫来抑制肿瘤生长,并克服对检查点阻断和 T 细胞介导的免疫疗法的治疗耐药性。
- 从机制上讲,肿瘤细胞和 cDC1 通过转运蛋白 SLC38A2 竞争谷氨酰胺的摄取,以调节抗肿瘤免疫。营养筛选和综合分析表明,谷氨酰胺是促进 cDC1 功能的主要氨基酸。此外,通过 FLCN 的谷氨酰胺信号传导会影响 TFEB 功能。 DC 中 FLCN 的缺失会以 TFEB 依赖性方式选择性损害体内 cDC1 功能,并通过消除补充谷氨酰胺的抗肿瘤治疗作用来表现 SLC38A2 缺陷。
- 我们的研究结果建立了肿瘤细胞和 cDC1 之间谷氨酰胺介导的细胞间代谢串扰,从而支持肿瘤免疫逃避,并揭示了 cDC1 中谷氨酰胺的获取和信号传导作为 DC 激活的限制事件和癌症治疗的假定靶标。
丙型肝炎病毒 RNA 5′-端有黄素腺嘌呤二核苷酸帽
Hepatitis C virus RNA is 5′-capped with flavin adenine dinucleotide. Nature
- RNA 病毒已经进化出复杂的策略来保护其基因组,包括 5′ 加帽。然而,迄今为止,尚未发现丙型肝炎病毒 (HCV) 的 RNA 5′ 帽,丙型肝炎病毒会导致慢性感染、肝硬化和癌症。
- 在这里,我们证明细胞代谢物黄素腺嘌呤二核苷酸 (FAD) 被病毒 RNA 依赖性 RNA 聚合酶用作非规范起始核苷酸,从而在 HCV RNA 上形成 5′-FAD 帽。 HCV FAD 封顶频率约为 75%,这是在所有生命王国中观察到的任何 RNA 代谢物封顶频率中最高的。 FAD 加帽在 HCV 分离株中对于复制中间负链和部分正链是保守的。在体内从患者样本以及人肝嵌合小鼠模型的肝脏和血清中分离的 HCV RNA 上也观察到了这一点。
- 此外,我们发现 5′-FAD 加帽可以保护 RNA 免受 RIG-I 介导的先天免疫识别,但不能稳定 HCV RNA。
- 这些结果将细胞代谢物加帽确立为一种新型病毒 RNA 加帽策略,可被其他病毒使用并影响抗病毒治疗结果和感染持续性。
bHLH 转录因子和组蛋白之间的合作以获取 DNA
Cooperation between bHLH transcription factors and histones for DNA access. Nature
- 转录因子的基本螺旋-环-螺旋 (bHLH) 家族可识别称为 E-box (CANNTG) 的 DNA 基序,包括 108 个成员。在这里,我们研究了染色质化 E-box 如何与两种结构不同的 bHLH 蛋白结合:原癌基因 MYC-MAX 和昼夜节律转录因子 CLOCK-BMAL1。
- 两种转录因子均优先与核小体入口-出口位点附近的 E-box 结合。对工程或天然核小体序列的结构研究表明,MYC-MAX 或 CLOCK-BMAL1 会触发组蛋白释放 DNA 以获取访问权限。在 H2A–H2B 酸性补丁顶部,CLOCK-BMAL1 Per-Arnt-Sim (PAS) 二聚化结构域与组蛋白八聚体盘结合。串联 E-boxes在内源 DNA 序列上的结合是通过两个 CLOCK-BMAL1 原聚体和组蛋白之间的直接相互作用发生的,对于昼夜节律循环非常重要。在内部 E-box 中,MYC-MAX 亮氨酸拉链还可以与组蛋白 H2B 和 H3 相互作用,并且其结合通过核小体上其他位置的 OCT4 间接增强。核小体E-box的位置和bHLH二聚化结构域的类型共同决定了组蛋白的接触、亲和力以及与其他核小体结合因子的竞争和协同程度。
治疗诱导的 APOBEC3A 驱动持久性癌细胞的进化
Therapy-induced APOBEC3A drives evolution of persistent cancer cells. Nature
看摘要有点水
- 抗癌靶向治疗的获得性耐药仍然是一个未解决的临床问题。尽管已经确定了获得性耐药的许多驱动因素,但治疗过程中影响肿瘤进化的潜在分子机制尚不完全清楚。患者肿瘤的基因组分析表明载脂蛋白 B 信使 RNA 编辑催化多肽样 (APOBEC) 胞苷脱氨酶与肿瘤进化有关;然而,它们在治疗和获得性耐药性发展过程中的作用尚不清楚。
- 在此,我们报道临床常用的肺癌靶向治疗可以诱导胞苷脱氨酶 APOBEC3A (A3A),导致治疗期间耐药癌细胞持续发生持续突变。治疗诱导的 A3A 促进双链 DNA 断裂的形成,增加耐药持久者的基因组不稳定性。 A3A 的缺失可减少持久细胞中的 APOBEC 突变和结构变异,并延缓耐药性的发展。 APOBEC 突变特征在肺癌患者的肿瘤中丰富,这些患者在对靶向治疗产生长期反应后病情进展。
- 这项研究表明,针对靶向治疗的诱导 A3A 驱动了耐药持久细胞的进化,这表明抑制 A3A 表达或活性可能是预防或延迟肺癌靶向治疗获得性耐药的潜在治疗策略。
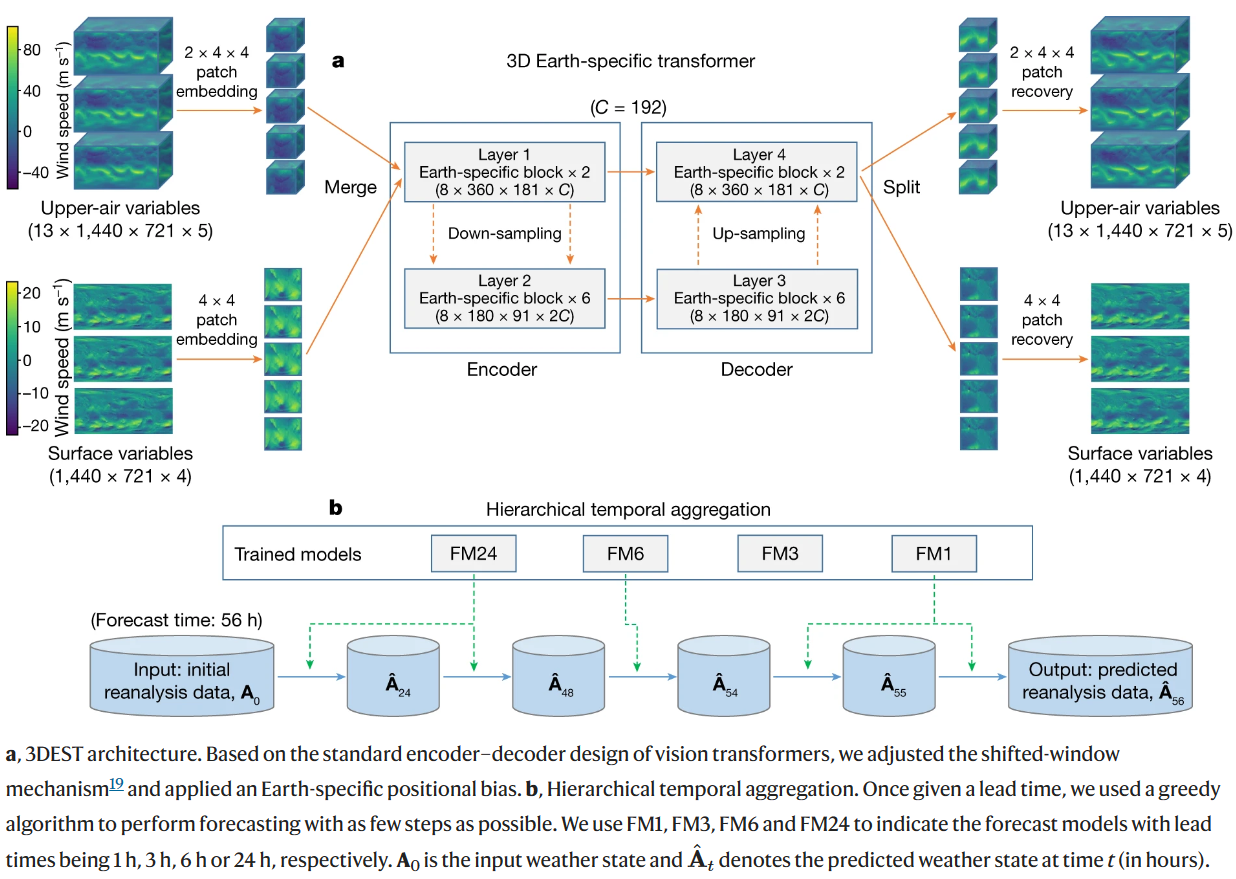
(~ ̄▽ ̄)~利用 3D 神经网络进行准确的中期全球天气预报
Accurate medium-range global weather forecasting with 3D neural networks. Nature
比较出圈的研究(本科毕业生一作发Nature,独立完成9成工作量)。有许多文献解读,比如:华为云提出盘古气象大模型:中长期气象预报精度首次超过传统数值方法,速度提升10000倍以上 – 知乎。
- 天气预报对于科学和社会都很重要。目前,最准确的预报系统是数值天气预报(NWP)方法,该方法将大气状态表示为离散网格,并数值求解描述这些状态之间转变的偏微分方程。然而,这个过程的计算成本很高。最近,基于人工智能的方法显示出将天气预报加速几个数量级的潜力,但预报精度仍然明显低于数值天气预报方法。
- 在这里,我们介绍一种基于人工智能的准确、中期全球天气预报的方法。我们证明,配备地球特定先验的三维深度网络可以有效处理天气数据中的复杂模式,并且分层时间聚合策略可以减少中期预测中的累积误差。
- 我们的盘古天气程序经过 39 年的全球数据训练,与世界上最好的 NWP 系统(欧洲中期天气中心的业务综合预报系统)相比,对所有测试变量的再分析数据获得了更强的确定性预报结果预测(ECMWF)。我们的方法也适用于极端天气预报和集合预报。当用再分析数据初始化时,跟踪热带气旋的精度也高于ECMWF-HRES。
- 摘录自此文:为了缓解迭代误差,本文提出一个简单而有效的策略。研究人员训练了4个不同预报间隔的模型,分别为1小时间隔、3小时间隔、6小时间隔、24小时间隔。进而,研究人员使用贪心算法调用这些模型,使得预测特定时间气象状况的迭代次数最小。
肿瘤免疫类
CD39 耗竭的工程 T 细胞治疗结直肠癌及其肝转移
CD39其实算是一个老靶点
- 结直肠肿瘤通常被免疫细胞密集浸润,这些细胞在监视和调节肿瘤进展中发挥作用,但受到免疫抑制信号的影响,从原发期到转移期,免疫抑制信号可能有所不同。在这里,我们采用多维方法来揭示原发性结直肠癌 (CRC) 和肝转移中的 T 细胞功能景观,并使用基因组编辑工具来开发 CRC 特异性工程化 T 细胞。
- 我们结合高维流式细胞术、RNA 测序和免疫组织化学来描述原发性和转移性 CRC 患者健康和肿瘤组织中 T 细胞的功能表型,并应用慢病毒载体 (LV) 和 CRISPR/Cas9 基因组编辑技术开发 CRC 专用细胞产品。
- 我们发现 T 细胞主要位于前缘,肿瘤浸润 T 细胞共表达多种抑制性受体,这些受体从原发部位到转移部位有很大差异。我们的数据强调 CD39 是原发性和转移性结直肠肿瘤衰竭的主要驱动因素。因此,我们同时采用一种针对 HER-2 的新型 T 细胞受体重定向 T 细胞特异性,并破坏内源 TCR 基因(TCR 编辑 (TCRED))和 CD39 编码基因 (ENTPD1),从而生成 TCREDENTPD1KOHER-2 重定向淋巴细胞。我们发现,CD39 的缺失赋予 HER-2 特异性 T 细胞在体外和体内消除 HER-2+ 患者来源的类器官方面的功能优势。
- HER-2 特异性 CD39 破坏的工程 T 细胞是治疗原发性和转移性 CRC 的有前途的先进医药产品。
新抗原特异性干细胞记忆样CD4+ T细胞介导MHC II类阴性实体瘤的CD8+ T细胞依赖性免疫治疗
- CD4+ T 细胞在一系列免疫反应中发挥着关键作用,无论是作为直接效应器还是通过辅助细胞(包括 CD8+ T 淋巴细胞)。在癌症中,能够直接识别肿瘤的新抗原 (NeoAg) 特异性 CD8+ T 细胞已被广泛研究,而 NeoAg 特异性 CD4+ T 细胞的作用尚不清楚。我们已经在单 T 细胞受体 (TCR) 克隆型水平和过继免疫治疗的设置。
- 我们发现天然的 CLTCH129>Q特异性库是多种多样的,并且包含通过四聚体结合测定和 CD4 依赖性测量的具有不同亲合力的 TCR。尽管存在这些差异,表达高或中等亲和力 TCR 的 CD4+ T 细胞在体内增殖能力与来自生长肿瘤的交叉呈递抗原相当,并驱动相似水平的依赖于 CD8+ T 细胞和 CD40L 信号传导的治疗免疫。
- 当 TCR 工程细胞用 IL-7 和 IL-15 而不是 IL-2 离体分化时,使用 NeoAg 特异性 CD4+ T 细胞的过继细胞疗法 (ACT) 最有效,这与扩增增加以及细胞增殖增加相关。在肿瘤引流淋巴结(tdLN)中获得并稳定维持 T 干细胞记忆(TSCM)样表型。使用 TSCM 样 CD4+ T 细胞进行 ACT 会降低肿瘤微环境中 CD8+ T 细胞的 PD-1 表达,并增加 tdLN 中 PD-1+CD8+ T 细胞的频率。
- 这些发现阐明了 NeoAg 特异性 CD4+ T 细胞通过为 CD8+ T 细胞提供帮助来介导抗肿瘤免疫的作用,并强调了它们在 ACT 中的治疗潜力。
(~ ̄▽ ̄)~ 多价RTK抑制剂增强免疫贫瘠型胃癌对ICI的反应
Multivalent tyrosine kinase inhibition promotes T cell recruitment to immune-desert gastric cancers by restricting epithelial-mesenchymal transition via tumour-intrinsic IFN-γ signalling. Gut. full pdf
其研究发现和PAD-GSClassifier有重叠之处。已加入PAD分型讨论中。
了解GC分类的算法
- 目的:胃癌(GC)在全球发病率中排名第五,死亡率排名第四。由于肿瘤固有的和获得性免疫治疗耐药性,GC 对免疫检查点阻断 (ICB) 治疗的反应是异质的。我们根据免疫细胞浸润开发了一种基于免疫表型的人类GC亚型,以开发一种新的治疗方案。
- 设计:开发了一种算法将GC重新分类为免疫炎症、排除和沙漠亚型。利用生物信息学、人和小鼠GC细胞系、同基因小鼠胃肿瘤模型和CTLA4阻断来研究通过限制受体酪氨酸激酶(RTK)信号传导在免疫沙漠(ICB抗性)型GC中的免疫治疗效果。
- 我们的算法对公共数据库中人类GC的亚型进行了重新分层,结果表明,与免疫炎症GC相比,免疫沙漠型和排除型肿瘤具有ICB耐药性。此外,上皮间质转化(EMT)信号在免疫沙漠型GC中高度富集,并且与上皮样相比,同基因小鼠肿瘤表现出间质样特性,具有T细胞排斥性并且对CTLA4阻断具有抵抗力。
- 我们的分析进一步确定了一组 RTK 作为免疫沙漠型 GC 中潜在的可药物靶标。 Dovitinib 是多种 RTK 的抑制剂,在类间充质免疫沙漠同基因 GC 模型中显着抑制 EMT 编程。 Dovitinib 激活肿瘤固有的 SNAI1/2-IFN-γ 信号轴并阻碍 EMT 程序,将免疫沙漠型肿瘤转化为免疫炎症型肿瘤,使这些间质样“冷”肿瘤对 CTLA4 阻断敏感。
- 结论:我们的研究结果确定了与患者群体相关的潜在药物靶标,特别是对于难治性免疫沙漠型/“冷”GC。 RTK 抑制剂 Dovitinib 通过限制 EMT 和招募 T 细胞,使沙漠型免疫冷 GC 对 CTLA4 阻断敏感。
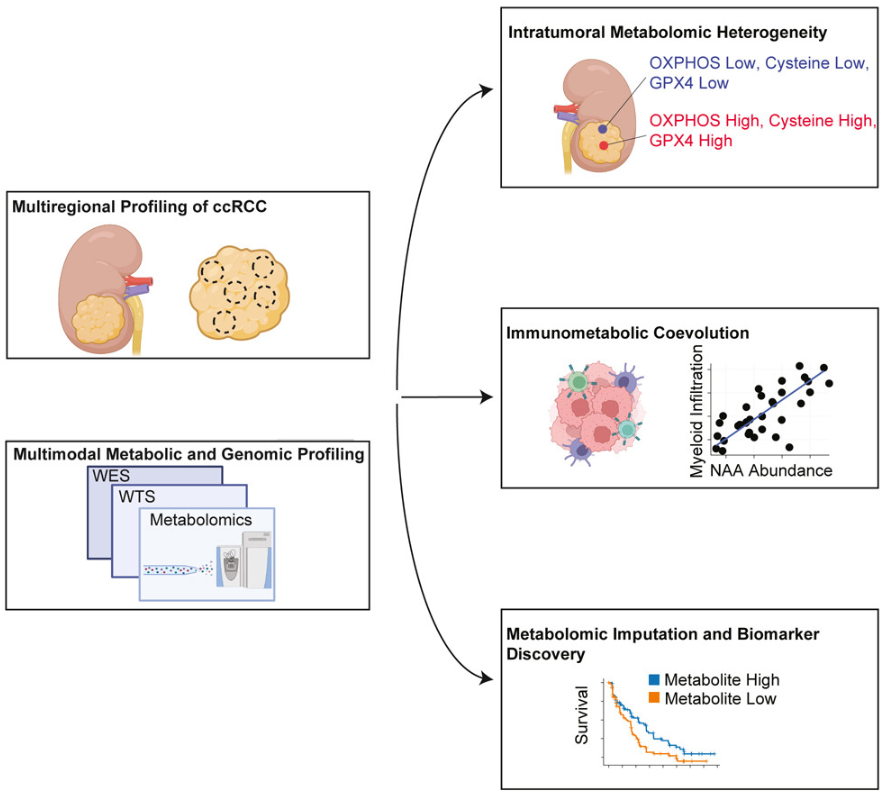
(~ ̄▽ ̄)~ 免疫代谢协同进化定义了 ccRCC 中独特的微环境生态位
Immunometabolic coevolution defines unique microenvironmental niches in ccRCC. Cell Metabolism. full html
了解一下如何从RNA-Seq推断代谢组图谱
- 肿瘤细胞表型和抗肿瘤免疫反应由局部代谢物可用性决定,但肿瘤内代谢物异质性 (IMH) 及其表型后果仍知之甚少。
- 为了研究 IMH,我们对透明细胞肾细胞癌 (ccRCC) 患者的肿瘤/正常区域进行了分析。 IMH 的一种常见模式在所有患者中均可见,其特征是与铁死亡相关的代谢物丰度和过程的相关波动。
- 瘤内代谢物-RNA 共变分析表明,微环境的免疫组成,尤其是骨髓细胞的丰度,驱动了瘤内代谢物的变化。受 RNA 代谢物共变的强度和 ccRCC 中 RNA 生物标志物的临床意义的启发,我们从参加 7 项临床试验的 ccRCC 患者的 RNA 测序数据中推断出代谢组图谱,并最终确定了与抗血管生成反应相关的代谢物生物标志物代理。
- 因此,局部代谢表型与免疫微环境同时出现,影响肿瘤的持续进化,并与治疗敏感性相关。
- Bensz摘录:we reconstructed data-modality-specific evolutionary phylogenies for each patient with at least 3 tumor regions profiled (n = 23 total)
将肿瘤细胞重编程为专业抗原提呈细胞
Restoring tumor immunogenicity with dendritic cell reprogramming. Sci Immunol
听上去略显疯狂的研究
- 抗原呈递减少有助于癌细胞逃避免疫系统的能力。我们使用 1 型常规树突状细胞 (cDC1) 的最小基因调控网络将癌细胞重编程为专业抗原呈递细胞 (肿瘤 APC)。
- 转录因子 PU.1、IRF8 和 BATF3 (PIB) 的强制表达足以在源自人类和小鼠血液肿瘤和实体瘤的 36 个细胞系中诱导 cDC1 表型。重编程后 9 天内,肿瘤 APC 获得了与 cDC1 细胞相关的转录和表观遗传程序。重编程恢复了肿瘤细胞表面抗原呈递复合物和共刺激分子的表达,允许内源性肿瘤抗原呈递在MHC-I上,并促进CD8+ T细胞的靶向杀伤。
- 从功能上讲,肿瘤 APC 吞噬并加工蛋白质和死亡细胞,分泌炎症细胞因子,并将抗原交叉呈递给初始 CD8+ T 细胞。人类原发性肿瘤细胞也可以被重新编程,以增强其呈递抗原和激活患者特异性肿瘤浸润淋巴细胞的能力。除了获得改善的抗原呈递之外,肿瘤 APC 还损害了体外和体内的致瘤性。将体外生成的黑色素瘤来源的肿瘤 APC 注射到皮下黑色素瘤肿瘤中,可以延迟肿瘤生长并提高小鼠的存活率。肿瘤 APC 引发的抗肿瘤免疫与免疫检查点抑制剂具有协同作用。
- 我们的方法作为开发免疫疗法的平台,赋予癌细胞处理和呈递内源性肿瘤抗原的能力。
临床类
唾液细胞外 miRNA 用于食管癌早期检测和预后的临床研究
Salivary extracellular miRNAs for early detection and prognostication of esophageal cancer: a clinical study. Gastroenterology
- 背景和目的:早期发现食管鳞状细胞癌(ESCC)将有助于治疗。我们的目标是建立源自唾液细胞外囊泡和颗粒 (EVP) 的微小 RNA (miRNA) 特征,用于早期 ESCC 检测和预测。
- 方法:使用微阵列对试点队列 (n=54) 中的唾液 EVP miRNA 表达进行分析。使用接受者-操作者特征曲线下面积 (AUROC) 和套索回归分析来确定区分 ESCC 患者和对照的 miRNA 的优先级。使用定量逆转录聚合酶链式反应,对发现队列 (n=72) 和细胞系中的候选者进行测量。生物标志物的预测模型源自训练队列 (n=342),并在内部队列 (n=207) 和外部队列 (n=226) 中进行验证。
- 结果:微阵列分析鉴定出 7 个 miRNA,用于区分 ESCC 患者和对照受试者。由于其中一种在发现队列和细胞系中并不总是可检测到,因此其他 6 种 miRNA 形成了一个组。该小组的签名准确识别了训练队列中的所有阶段 ESCC 患者 (AUROC=0.968),并在两个独立队列中成功得到验证。重要的是,该特征可以区分早期(Ⅰ/Ⅱ期)ESCC患者与训练队列中的对照受试者(AUROC=0.969,敏感性=92.00%,特异性=89.17%),内部(敏感性=90.32%,特异性=91.04) %)和外部(敏感性=91.07%,特异性=88.06%)验证队列。此外,建立了基于该面板的预后特征,并有效预测了无进展生存期和总生存期较差的高风险病例。
- 结论:基于唾液 EVP 的 6-miRNA 特征可以作为 ESCC 早期检测和风险分层的非侵入性生物标志物。中国临床试验注册中心,ChiCTR2000031507。
Atezolizumab vs. 单药化疗治疗铂类不耐受性晚期NSCLC有显著获益
- 背景:尽管晚期或转移性非小细胞肺癌 (NSCLC) 患者的免疫治疗取得了进展,但关键的一线试验仅限于东部肿瘤合作组体能状态 (ECOG PS) 0-1 且中位年龄的患者65 岁或以下。我们的目的是比较一线 atezolizumab 单药治疗与单药化疗对不适合铂类化疗的患者的疗效和安全性。
- 方法:本试验是一项 3 期、开放标签、随机对照研究,在亚洲、欧洲、北美和南美 23 个国家的 91 个地点进行。符合条件的 IIIB 或 IV 期 NSCLC 患者,由于 ECOG PS 2 或 3,研究者认为不适合铂类双药化疗,或者年龄为 70 岁或以上,ECOG PS 0-1,且有严重合并症或禁忌症。铂类双药化疗。通过置换区组随机化(区组大小为 6 个)将患者按照 2:1 的比例随机分配,接受每 3 周静脉注射 1200 毫克阿替利珠单抗或单药化疗(长春瑞滨 [口服或静脉注射] 或吉西他滨 [静脉注射];按照当地标签给药),以 3 周或 4 周为周期。主要终点是意向治疗人群的总体生存率评估。安全性分析是在可安全评估的人群中进行的,其中包括接受任何剂量的阿替利珠单抗或化疗的所有随机患者。该试验已在 ClinicalTrials.gov 注册,NCT03191786。
- 结果:2017 年 9 月 11 日至 2019 年 9 月 23 日期间,453 名患者入组并随机接受阿特珠单抗 (n=302) 或化疗 (n=151)。与化疗相比,Atezolizumab 改善了总生存期(中位总生存期为 10·3 个月 [95% CI 9·4-11·9] vs 9·2 个月 [5·9-11·2];分层风险比 0·78 [0 ·63-0·97],p=0·028),阿特珠单抗组的 2 年生存率为 24% (95% CI 19·3-29·4),而化疗的 2 年生存率为 12% (6·7-18· 0) 。
- 与化疗相比,atezolizumab 与患者报告的健康相关生活质量功能量表和症状的稳定或改善相关,并且 3-4 级治疗相关不良事件较少(300 例中有 49 例 [16%] vs 49 例[33] %](占 147 例)和治疗相关死亡(三例 [1%] 对四例 [3%])。
- 解释:与单药化疗相比,阿替利珠单抗单药一线治疗可改善总生存率、使 2 年生存率加倍、维持生活质量以及良好的安全性。这些数据支持 atezolizumab 单药疗法作为不适合接受铂类化疗的晚期 NSCLC 患者的潜在一线治疗选择。
淋巴结阴性、雌激素受体阳性乳腺癌女性的基因表达和化疗的益处
Gene Expression and Benefit of Chemotherapy in Women With Node-Negative, Estrogen Receptor-Positive Breast Cancer. J Clin Oncol
- 21 基因复发评分 (RS) 测定量化了接受他莫昔芬辅助治疗的雌激素受体阳性、淋巴结阴性乳腺癌女性远处复发的可能性。 RS 与化疗获益之间的关系尚不清楚。
- 在国家乳腺和肠道辅助手术项目 (NSABP) B20 试验中,测量了他莫昔芬治疗和他莫昔芬联合化疗治疗患者肿瘤的 RS。 Cox 比例风险模型用于测试化疗治疗与 RS 之间的相互作用。
- 共有 651 名患者进行评估(227 名患者随机分配至他莫昔芬组,424 名患者随机分配至他莫昔芬联合化疗组)。化疗治疗与 RS 之间相互作用的检验具有统计学显着性 (P = .038)。高RS(≥ 31)肿瘤(即高复发风险)患者从化疗中获益较大(相对风险,0.26;95% CI,0.13至0.53;10年远处复发率绝对降低:平均, 27.6%;SE,8.0%)。低 RS (< 18) 肿瘤患者从化疗中获益甚微(如果有的话)(相对风险,1.31;95% CI,0.46 至 3.78;10 年远处复发率绝对降低:平均值,-1.1%;东南欧,2.2%)。患有中等 RS 肿瘤的患者似乎没有获得很大的获益,但估计的不确定性不能排除临床上重要的获益。
- 总之,RS 测定不仅可以量化淋巴结阴性、雌激素受体阳性乳腺癌女性乳腺癌复发的可能性,还可以预测化疗获益的程度。
体内长寿CAR-T细胞的转录组特征
Transcriptional signatures associated with persisting CD19 CAR-T cells in children with leukemia. Nat Med
- 在复发难治性儿童前 B 细胞急性淋巴细胞白血病 (R/R B-ALL) 的背景下,靶向 CD19 的嵌合抗原受体 (CAR)-T 细胞通常会诱导持久缓解,这需要 CAR-T 细胞的持续存在。
- 在本研究中,我们通过高通量单细胞基因表达和输液产品以及连续血液和骨髓的 T 细胞受体测序,系统分析了参加 CARPALL 试验的 10 名 R/R B-ALL 儿童的 CD19 CAR-T 细胞输注后 5 年内可取样。
- 我们发现,长寿CAR-T 细胞形成了 CD4/CD8 双阴性表型,具有类似耗尽的记忆状态和独特的转录特征。这种持久性特征在所有具有长期治疗反应的儿童的循环 CAR-T 细胞中占主导地位,对此测序数据已足够(4/4,100%)。该特征也存在于 T 细胞亚群和克隆型中,表明持续存在的 CAR-T 细胞在转录上趋于一致。这种持久性特征也在两名接受不同 CD19 CAR-T 细胞产品且缓解长达十年的慢性淋巴细胞白血病成年患者中检测到。对儿童和成人各种健康和患病组织的单 T 细胞转录组的检查表明,持久性特征可能是长寿命 CAR-T 细胞所特有的。
- 这些发现提出了临床有效、持久的 CD19 CAR-T 细胞存在通用转录特征的可能性。
慢性疼痛发生和扩散的预后风险评分
A prognostic risk score for development and spread of chronic pain. Nat Med
- 慢性疼痛是一种复杂的疾病,受生物、心理和社会因素的综合影响。
- 利用英国生物银行 (n = 493,211) 的数据,我们发现疼痛从近端部位扩散到远端部位,并开发了一种生物心理社会模型来预测共存疼痛部位的数量。该数据驱动模型用于确定对各种慢性疼痛状况(曲线下面积 (AUC) 0.70-0.88)和疼痛相关医疗状况(AUC 0.67-0.86)进行分类的风险评分。
- 在纵向分析中,风险评分预测了大约 9 年后广泛慢性疼痛的发展、慢性疼痛在身体部位的扩散以及高冲击性疼痛(AUC 0.68-0.78)。主要风险因素包括失眠、感到“厌倦”、疲劳、压力性生活事件和体重指数>30。该评分的简化版本称为疼痛扩散风险,基于六个简单问题和二值化答案获得了类似的预测性能。随后在芬兰北部出生队列 (n = 5,525) 和 PREVENT-AD 队列 (n = 178) 中验证了疼痛扩散的风险,获得了可比较的预测性能。
- 我们的研究结果表明,慢性疼痛状况可以通过一组常见的生物心理社会因素来预测,这有助于制定研究方案、优化临床试验中的患者随机化和改善疼痛管理。
粪便微生物群移植 + anti-PD-1治疗晚期黑色素瘤的安全性评估
Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat Med
怎样才可以成为FMT样本的捐赠者?他们与普通人的特征有哪些异同?
- 粪便微生物群移植(FMT)是克服难治性黑色素瘤患者对免疫检查点抑制剂耐药性的潜在策略;然而,FMT 在一线治疗中的作用尚未得到评估。
- 我们对 20 名未经治疗的晚期黑色素瘤患者进行了一项多中心 I 期试验,将健康供体 FMT 与 PD-1 抑制剂纳武单抗 (nivolumab) 或派姆单抗 (pembrolizumab) 相结合。主要终点是安全性。
- 仅 FMT 未报告 3 级不良事件。 5 名患者 (25%) 因联合治疗出现了 3 级免疫相关不良事件。关键的次要终点是客观缓解率、肠道微生物组组成的变化以及全身免疫和代谢组学分析。客观答复率为 65%(20 份中有 13 份),其中有 4 份(20%)完整答复。纵向微生物组分析显示,所有患者都移植了来自各自捐赠者的菌株;然而,在响应者中,捐赠者和患者微生物组之间获得的相似性只会随着时间的推移而增加。 FMT 后,应答者经历了免疫原性的富集和有害细菌的减少。 Avatar 小鼠模型证实了健康供体粪便在提高抗 PD-1 功效方面的作用。
- 我们的结果表明,来自健康捐赠者的 FMT 在一线环境中是安全的,值得与免疫检查点抑制剂结合进行进一步研究。 ClinicalTrials.gov 标识符 NCT03772899 。
其它类
人内源性逆转录病毒 K 与胶质母细胞瘤中的干细胞生态位
Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J Clin Invest
- 人类内源性逆转录病毒 (HERV) 是祖先病毒的遗迹,占人类基因组的近 8%。尽管通常处于沉默状态,但最近整合的原病毒 HERV-K (HML-2) 可以在某些癌症中重新激活。
- 在这里,我们报告了脑脊液和肿瘤组织中恶性胶质瘤中 HML-2 的病理表达,这与癌症干细胞表型和不良预后相关。
- 使用单细胞 RNA-Seq,我们鉴定了神经祖细胞样细胞 (NPC-like) 中 HML-2 转录物升高的胶质母细胞瘤细胞群,这些细胞可驱动细胞可塑性。使用 CRISPR 干扰,我们证明 HML-2 在胶质母细胞瘤神经球和颅内原位小鼠模型中关键维持胶质母细胞瘤干性和肿瘤发生。此外,我们证明 HML-2 严格调节 NPC 衍生的星形胶质细胞中的胚胎干细胞程序,并通过激活核转录因子 OCT4 改变其 3D 细胞形态,OCT4 与 HML-2 特异性长末端重复序列 (LTR5Hs) 结合。
- 此外,我们发现一些胶质母细胞瘤细胞形成不成熟的逆转录病毒颗粒,用抗逆转录病毒药物抑制 HML-2 表达会降低细胞外区室中的逆转录酶活性、肿瘤活力和多能性。
- 我们的结果表明 HML-2 从根本上有助于胶质母细胞瘤干细胞生态位。由于胶质母细胞瘤干细胞的持续存在被认为是治疗耐药和复发的原因,因此 HML-2 可能作为独特的治疗靶点。
融合蛋白与肿瘤治疗
Dissolution of oncofusion transcription factor condensates for cancer therapy. Nat Chem Biol
- 癌症相关的染色体重排可导致多种致病性融合蛋白的表达。融合蛋白促进肿瘤发生的机制在很大程度上尚不清楚,并且缺乏针对融合相关癌症的有效疗法。在这里,我们全面检查了各种癌症中发现的融合蛋白。
- 我们发现许多融合蛋白由易相分离结构域(PS)和DNA结合结构域(DBD)组成,这些融合与异常基因表达模式有很强的相关性。
- 此外,我们建立了一种名为 DropScan 的高通量筛选方法来筛选能够调节异常冷凝物的药物。通过 DropScan 鉴定出的一种药物 LY2835219 可以有效溶解表达尤文肉瘤融合体的报告细胞系中的凝聚物,并部分挽救目标基因的异常表达。
- 我们的结果表明,异常相分离可能是这些 PS-DBD 融合相关癌症的常见机制,并表明调节异常相分离是治疗这些疾病的潜在途径。
致癌 K-Ras 抑制整体 miRNA 功能
- K-Ras 经常获得功能获得性突变(K-RasG12D 最常见),从而触发显着的转录组和蛋白质组变化,从而驱动肿瘤发生。然而,我们对致癌过程中 K-Ras 诱导的 microRNA (miRNA) 等转录后调节因子的失调知之甚少。
- 在这里,我们报告 K-RasG12D 促进 miRNA 活性的全局抑制,导致数百个靶标的上调。我们使用 Halo 增强的 Argonaute pull-down 构建了表达 K-RasG12D 的小鼠结肠上皮和肿瘤中生理 miRNA 靶标的全面概况。
- 将其与染色质可及性、转录组和蛋白质组的平行数据集相结合,我们发现 K-RasG12D 抑制 Csnk1a1 和 Csnk2a1 的表达,随后降低 Ago2 在 Ser825/829/832/835 处的磷酸化。低磷酸化的 Ago2 增加了与 mRNA 的结合,同时降低了其抑制 miRNA 靶点的活性。
- 我们的研究结果将病理生理学背景下的全局 miRNA 活性的有效调节机制与 K-Ras 联系起来,并提供致癌 K-Ras 与 miRNA 靶标转录后上调之间的机制联系。
G4access 识别 G-四链体及其与开放染色质和印记控制区域的关联
- 后生动物启动子富含二级 DNA 结构形成基序,例如 G-四链体 (G4s)。
- 在这里,我们描述了“G4access”,一种通过核酸酶消化分离和测序与开放染色质相关的 G4 的方法。 G4access 不依赖于抗体和交联,并且丰富了计算预测的 G4 (pG4),其中大部分在体外得到了证实。
- 在人和小鼠细胞中使用 G4access,我们鉴定了与核小体排除和启动子转录相关的细胞类型特异性 G4 富集。 G4access 允许测量 G4 配体处理、HDAC 和 G4 解旋酶抑制剂后 G4 库使用的变化。将 G4access 应用到来自小鼠杂交杂交的细胞表明 G4 在控制活性印记区域中发挥着作用。一致地,我们还观察到 G4access 峰未甲基化,而 pG4 的甲基化与 DNA 上的核小体重新定位相关。
- 总体而言,我们的研究为研究细胞动力学中的 G4 提供了一种新工具,并强调了它们与开放染色质、转录及其对 DNA 甲基化的拮抗作用的关联。
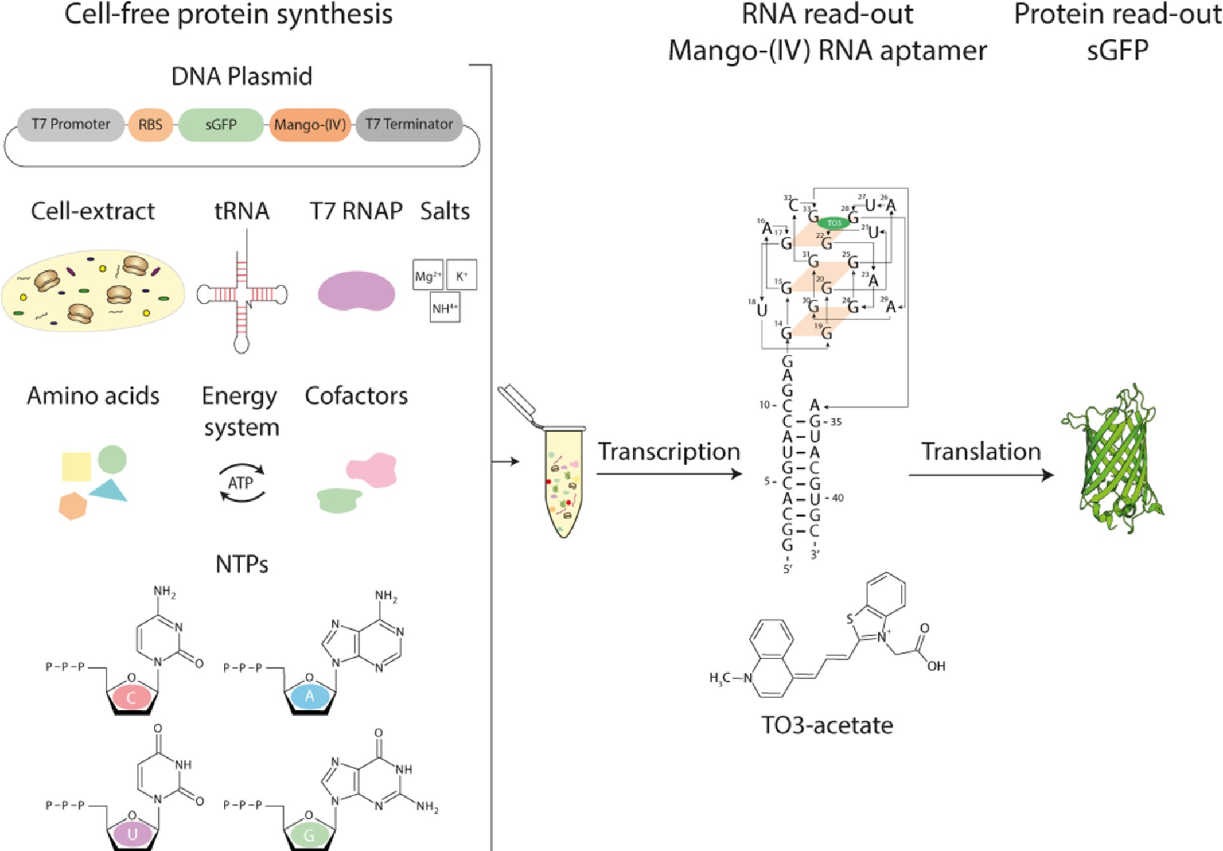
无细胞转录-翻译系统:表征核糖开关功能的双重读出测定
Cell-free transcription-translation system: a dual read-out assay to characterize riboswitch function. Nucleic Acids Res
- 无细胞蛋白质合成测定已成为了解转录和翻译过程的宝贵工具。
- 在这里,我们建立了一种基于荧光的体外转录-翻译耦合测定作为读出系统,以同时量化 mRNA 和蛋白质水平。
- 我们利用成熟的移位绿色荧光蛋白 (sGFP) 表达定量作为蛋白质水平的读数。此外,我们使用荧光 Mango-(IV) RNA 适体测定了 mRNA 量,该适体在与荧光团噻唑橙 (TO) 结合后发出荧光。我们利用由四个连续的 Mango-(IV) RNA 适体元件组成的 Mango-(IV) RNA 适体系统,通过构建 Mango 阵列提高了灵敏度。该报告基因测定的设计产生了具有高信噪比的灵敏读数,使我们能够通过连续监测荧光变化以及反应快照来监测无细胞测定中的转录和翻译时间过程。此外,我们应用这种双读出测定法来研究大肠杆菌的硫胺素感应核糖开关 thiM 和 thiC 以及创伤弧菌的腺嘌呤感应核糖开关 ASW 和枯草芽孢杆菌的 pbuE 的功能,它们代表转录和翻译的开启和关闭-分别是核糖开关。
- 这种方法实现了基于微孔板的应用,这是对核糖开关功能高通量筛选工具箱的一个有价值的补充。
使用深度学习预测 CRISPR-Cas13d 引导 RNA 的中靶和脱靶活性
Prediction of on-target and off-target activity of CRISPR-Cas13d guide RNAs using deep learning. Nat Biotechnol
- 转录组工程在具有 RNA 靶向 CRISPR 效应子的活细胞中的应用取决于对目标活性和脱靶避免的准确预测。
- 在这里,我们设计并测试了约 200,000 个 RfxCas13d 引导 RNA,这些 RNA 靶向人类细胞中的必需基因,并具有系统设计的错配以及插入和删除 (indel)。我们发现错配和插入缺失对 Cas13d 活性具有位置和上下文相关的影响,并且导致 G-U 摆动配对的错配比其他单碱基错配更容易容忍。
- 使用这个大规模数据集,我们训练了一个卷积神经网络,我们将其称为“通过 gRNA 设计靶向抑制基因表达”(TIGER),以根据引导序列和上下文预测功效。 TIGER 在预测我们的数据集和已发布数据集上的目标和脱靶活动方面优于现有模型。
- 我们证明,TIGER 评分与特定错配相结合产生了第一个调节转录表达的通用框架,从而能够使用 RNA 靶向 CRISPR 来精确控制基因剂量。
可穿戴运动追踪数据可在临床诊断前数年识别帕金森病
Wearable movement-tracking data identify Parkinson’s disease years before clinical diagnosis. Nat Med
腕戴式加速度计(wrist-worn accelerometry)?
- 帕金森病是一种进行性神经退行性运动障碍,具有较长的潜伏期,目前尚无缓解疾病的治疗方法。可以改变神经保护治疗开发工作的可靠预测生物标志物仍有待确定。
- 利用英国生物银行,我们研究了加速度测量法(accelerometry)在识别普通人群中前驱帕金森病方面的预测价值,并将这种数字生物标记物与基于遗传学、生活方式、血液生化或前驱症状数据的模型进行比较。
- 使用加速测量数据训练的机器学习模型在区分临床诊断的帕金森病 (n = 153)(精确回忆曲线下面积 (AUPRC) 0.14 ± 0.04)和前驱帕金森病 (n = 113) 方面达到了更好的测试性能,最多可达 7 年-一般人群(n = 33,009)的诊断(AUPRC 0.07 ± 0.03)与所有其他测试方式相比(遗传学:AUPRC = 0.01 ± 0.00,P = 2.2 × 10-3;生活方式:AUPRC = 0.03 ± 0.04,P = 2.5 × 10-3;血液生化:AUPRC = 0.01 ± 0.00,P = 4.1 × 10-3;前驱体征:AUPRC = 0.01 ± 0.00,P = 3.6 × 10-3)。
- 加速度测量是一种潜在重要的低成本筛查工具,可用于确定有患帕金森病风险的人,并确定神经保护治疗临床试验的参与者。
机器学习/组学类
人胃体上皮的空间解析表达景观和基因调控网络
Spatially resolved expression landscape and gene-regulatory network of human gastric corpus epithelium. Protein Cell. full html
All sequencing data generated in this study are deposited in the Genome Sequence Archive (GSA) database (accession number: HRA002917).
- 人类胃体上皮的分子知识仍然不完整。
- 在这里,通过使用单细胞 RNA 测序 (scRNA-seq)、空间转录组学和单细胞转座酶可及染色质测序 (scATAC-seq) 技术进行综合分析,我们揭示了空间分辨的表达景观和基因调控网络人胃体上皮。
- 具体来说,我们在人胃体峡部鉴定了干细胞/祖细胞群,其中 EGF 和 WNT 信号通路被激活。同时,LGR4而非LGR5负责WNT信号通路的激活。重要的是,FABP5 和 NME1 被鉴定和验证对于正常胃干/祖细胞和胃癌细胞都至关重要。最后,我们在染色质状态水平上探索了胃体上皮关键基因的表观遗传调控,并鉴定了几种重要的细胞类型特异性转录因子。
- 总之,我们的工作为系统地了解体内人胃体上皮的细胞多样性和稳态提供了新的见解。
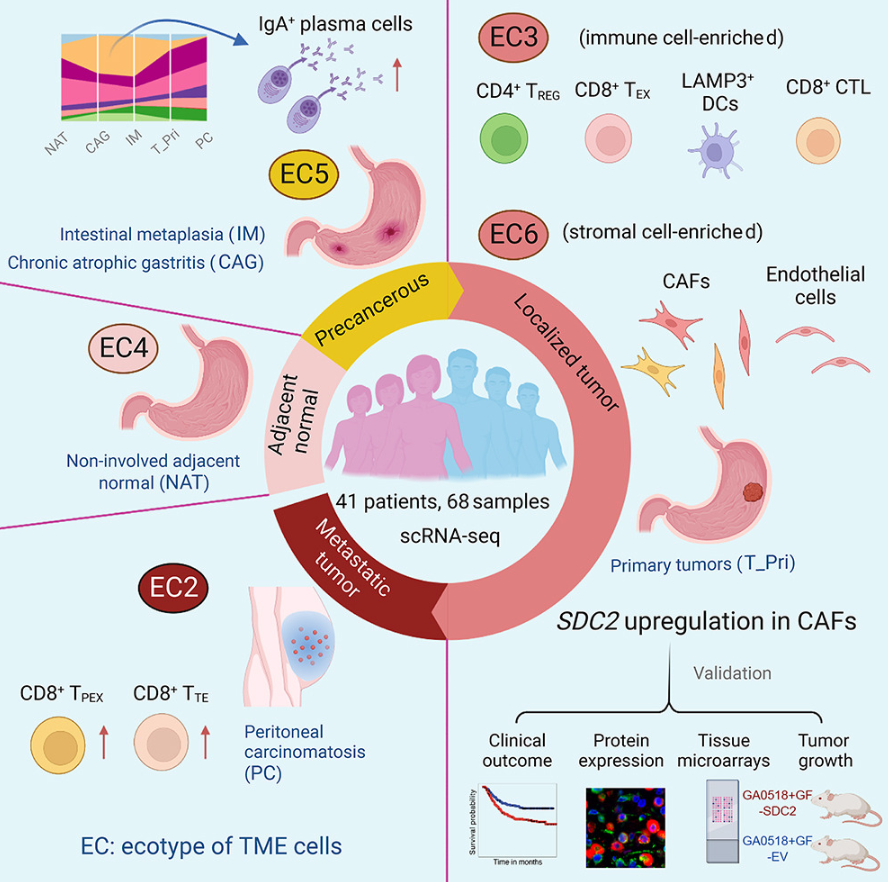
(~ ̄▽ ̄)~ 胃腺癌进展过程中免疫和基质细胞状态和生态型的演变
Evolution of immune and stromal cell states and ecotypes during gastric adenocarcinoma progression. Cancer Cell. full pdf
美国-德克萨斯大学 MD 安德森癌症中心-基因组医学系
了解一下数据
- 了解胃腺癌 (GAC) 进展中的肿瘤微环境 (TME) 重编程可能会发现新的治疗靶点。
- 在这里,我们对癌前病变、局部和转移性 GAC 进行了单细胞分析,识别了随着 GAC 进展 TME 细胞状态和成分的变化。癌前微环境中存在丰富的 IgA+ 浆细胞,而免疫抑制性骨髓和基质亚群在晚期 GAC 中占主导地位。
- 我们确定了六种 TME 生态类型 (EC1-6)。 EC1 是血液所独有的,而 EC4、EC5 和 EC2 分别在未受累组织、癌前病变和转移瘤中高度富集。 EC3 和 EC6 是原代 GAC 中两种不同的生态型,与组织病理学和基因组特征以及生存结果相关。
- GAC 进展过程中会发生广泛的基质重塑。癌症相关成纤维细胞 (CAF) 中 SDC2 的高表达与侵袭性表型和较差的生存率有关,而 CAF 中 SDC2 的过度表达会促进肿瘤生长。
- 我们的研究提供了高分辨率的 GAC TME 图集,并强调了进一步研究的潜在目标。
默克尔细胞癌异质性的单细胞解剖揭示了转录组可塑性和治疗脆弱性
- 默克尔细胞癌(Merkel cell carcinoma, MCC)是一种罕见但具有侵袭性的皮肤癌,在精准医疗时代仍然是一个挑战。免疫检查点抑制剂 (ICIs) 是唯一被批准用于治疗晚期 MCC 的疗法,但它受到高原发性和获得性耐药性的阻碍。
- 因此,我们以单细胞分辨率剖析了一组患者肿瘤中的转录组异质性,揭示了未接受治疗的 MCC 子集中的表型可塑性。处于“间充质样”状态的肿瘤细胞具有炎症表型,预示着更好的 ICI 反应。这一观察结果也在来自 MCC 患者肿瘤的最大的完整转录组数据集中得到了验证。相比之下,ICI 抗性肿瘤主要表达具有“免疫冷”景观的分化良好状态的神经上皮标志物。
- 重要的是,向“间充质样”状态的微妙转变(如何产生这种转变?)可恢复原代 MCC 细胞中的 copanlisib 耐药性,突显了患者分层治疗中利用肿瘤细胞可塑性、增强治疗功效和避免耐药性的潜在策略。
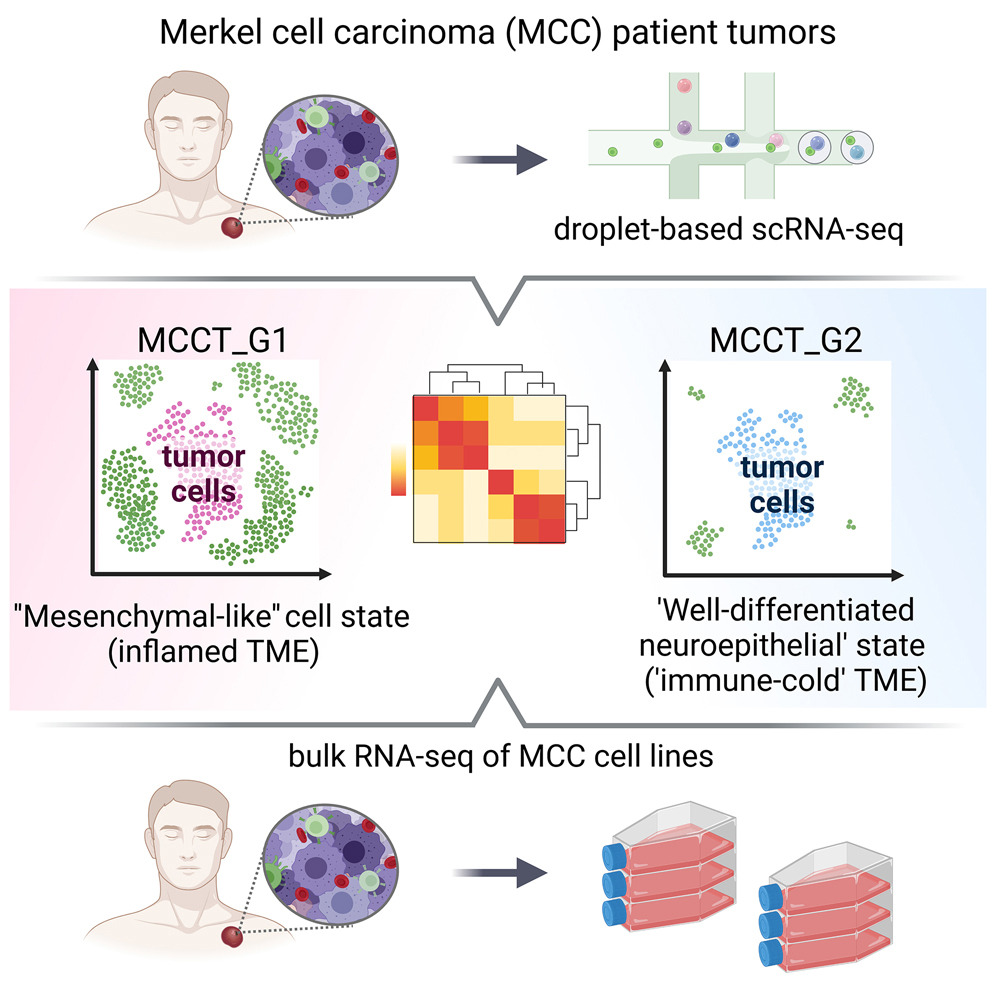
(~ ̄▽ ̄)~ MuAt:用于肿瘤分型和亚型分型的体细胞突变的深度表征学习
Mutation-Attention (MuAt): deep representation learning of somatic mutations for tumour typing and subtyping. Genome Med
芬兰赫尔辛基大学芬兰分子医学研究所 、赫尔辛基大学医学院应用肿瘤基因组学研究项目、3iCAN 数字精准癌症医学旗舰店
对GSClassifier有一定启示
英国基因组学研究联盟
- 背景:癌症基因组测序能够准确分类肿瘤和肿瘤亚型。然而,使用仅外显子组测序和体细胞突变负担较低的肿瘤类型(例如许多儿科肿瘤)的预测性能仍然有限。此外,利用深度表示学习来发现肿瘤实体的能力仍然未知(其实也不尽然)。
- 方法:我们在这里介绍突变注意力(MuAt),这是一种深度神经网络,用于学习简单和复杂的体细胞改变的表示,以预测肿瘤类型和亚型。与许多以前的方法相比,MuAt 利用个体突变的注意力机制而不是聚合突变计数。
- 我们使用来自全基因组泛癌症分析 (PCAWG) 的 2587 个完整癌症基因组(24 种肿瘤类型)和来自癌症基因组图谱 (TCGA) 的 7352 个癌症外显子组(20 种类型)训练 MuAt 模型。 MuAt 的全基因组预测准确率为 89%,全外显子组预测准确率为 64%,top-5 准确率分别为 97% 和 90%。 MuAt 模型经过良好校准,在三个独立的全癌症基因组队列(总共 10,361 个肿瘤)中表现良好。
- 我们证明 MuAt 能够学习临床和生物学相关的肿瘤实体,包括肢端黑色素瘤、SHH 激活的髓母细胞瘤、SPOP 相关的前列腺癌、微卫星不稳定性、POLE 校对缺陷和 MUTYH 相关的胰腺内分泌肿瘤,而无需这些肿瘤亚型和亚组作为培训标签提供。最后,对 MuAt 注意力矩阵的仔细研究揭示了简单和复杂体细胞突变的普遍存在和肿瘤类型特定模式。
- 结论:MuAt 学到的体细胞改变的综合表征能够准确识别组织学肿瘤类型并识别肿瘤实体,有可能影响精准癌症医学。
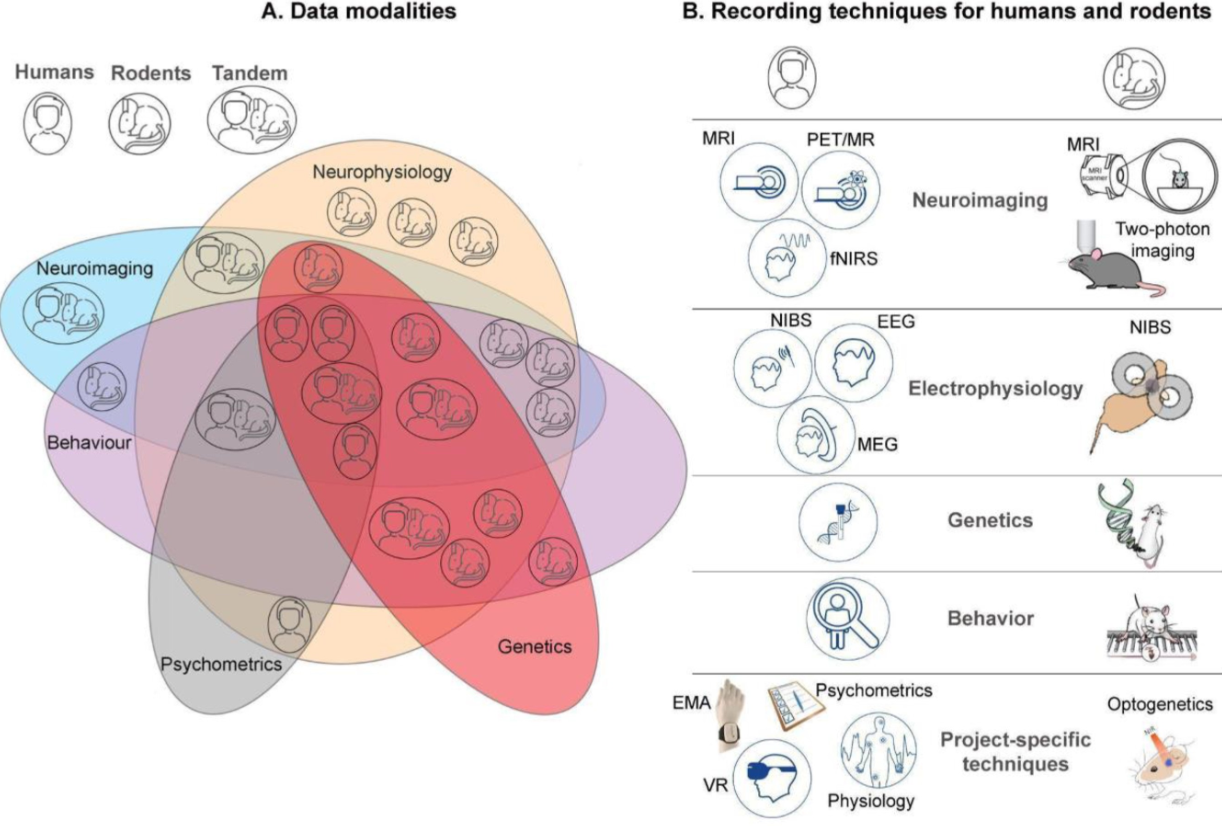
(~ ̄▽ ̄)~ 协作研究中心的数据管理策略
Data management strategy for a collaborative research center. Gigascience
有空可了解一下
- 随着数据采集技术和研究方法的每一次进步,有效的研究数据管理 (RDM) 策略对于支持可查找、可访问、可互操作和可重用 (FAIR) 神经科学数据的生成的重要性日益增长。为了最大限度地发挥多样化研究策略的影响,多学科、大规模的神经科学研究联盟在 RDM 中面临着许多未解决的挑战。尽管开放科学原则已被广泛接受,但研究人员实际上很难将 RDM 置于其他紧迫需求之上。为涵盖动物、人类和临床研究的联盟实施连贯、可执行的 RDM 计划变得越来越具有挑战性。
- 在这里,我们提出了为海德堡合作研究联盟实施的 RDM 策略。我们的联盟结合了不同人群(动物和人类)的基础和临床研究,并产生高度异质和多模式的研究数据(例如神经生理学、神经影像学、遗传学、行为)。
- 我们提出了一项为大型合作研究联盟启动早期 RDM 和 FAIR 数据生成的具体策略,重点是可持续的解决方案,激励增量 RDM,同时尊重研究的特定要求。
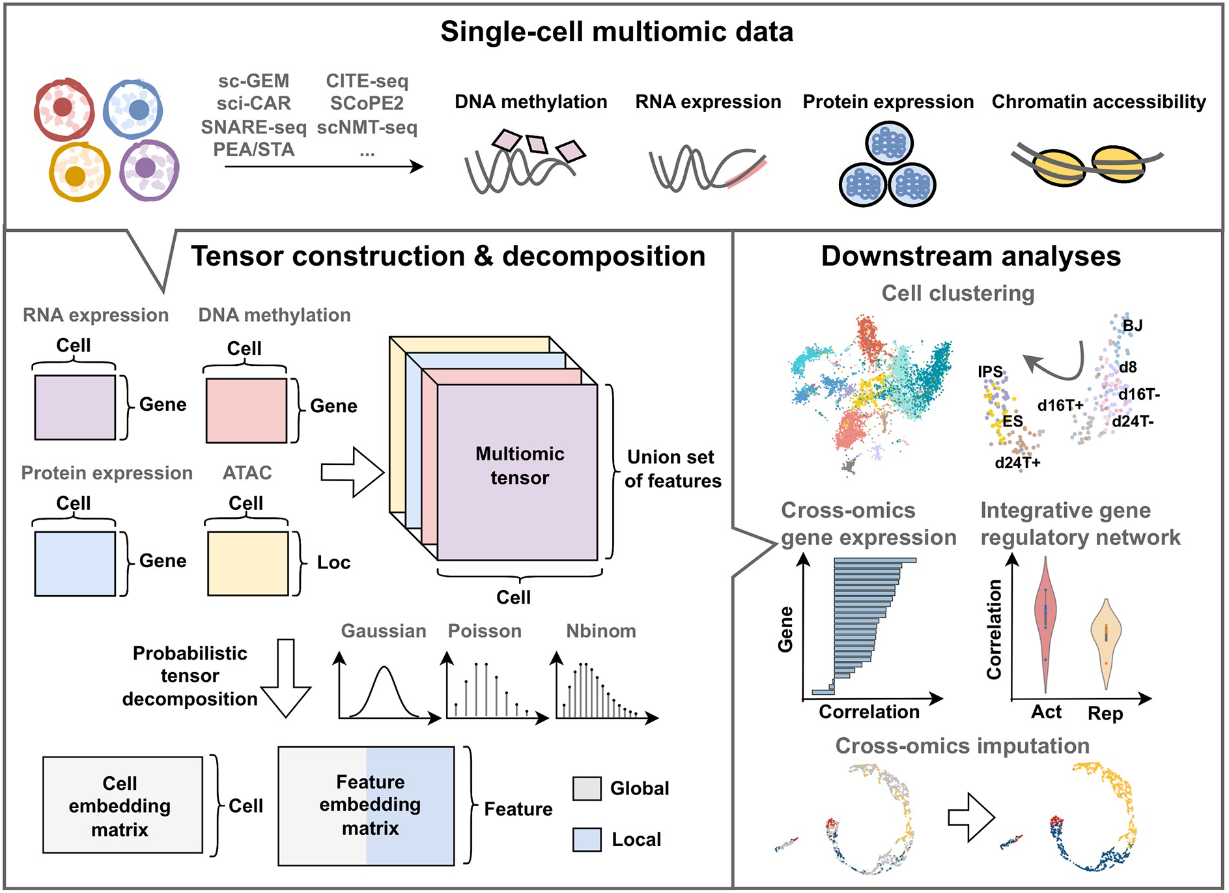
概率张量分解从单细胞多组学数据中提取更好的潜在嵌入
Probabilistic tensor decomposition extracts better latent embeddings from single-cell multiomic data. Nucleic Acids Res
香港城市大学计算机科学系
- 单细胞测序技术能够同时捕获多个细胞的多组学数据。捕获的数据可以用张量表示,即高阶矩阵。然而,现有的分析工具往往将数据视为二阶矩阵的集合,放弃了特征之间的对应关系。
- 因此,我们提出了一种概率张量分解框架 SCOIT,用于从单细胞多组学数据中提取嵌入。 SCOIT 结合了各种分布,包括高斯分布、泊松分布和负二项分布,来处理稀疏、噪声和异构的单细胞数据。我们的框架可以将多组学张量分解为细胞嵌入矩阵、基因嵌入矩阵和组学嵌入矩阵,从而允许进行各种下游分析。
- 我们将 SCOIT 应用于来自不同测序方案的八个单细胞多组学数据集。通过细胞嵌入,与各种指标下的九种最先进的工具相比,SCOIT 在细胞聚类方面实现了卓越的性能,证明了其剖析细胞异质性的能力。通过基因嵌入,SCOIT 能够实现跨组学基因表达分析和综合基因调控网络研究。此外,嵌入允许同时进行跨组学插补,优于当前的插补方法,皮尔逊相关系数增加了 3.38-39.26%;此外,SCOIT 适应细胞子集仅具有一个可用的组学特征的情况。
基于多源特征和深度学习预测潜在的微生物与疾病关联
Predicting potential microbe-disease associations based on multi-source features and deep learning. Brief Bioinform
- 研究证实,人体内许多复杂疾病的发生与微生物群落密切相关,微生物可以通过调节肿瘤微环境影响肿瘤的发生和转移。然而,疾病微生物群的临床观察仍然存在很大差距。尽管生物实验可以准确地识别与疾病相关的微生物,但它们也既耗时又昂贵。有效识别疾病相关微生物的计算模型可以缩短这一过程,并降低资金和时间成本。
- 基于此,本文提出了一种名为 DSAE_RF 的模型,通过结合多源特征和深度学习来预测潜在的微生物与疾病的关联。 DSAE_RF 计算微生物和疾病之间的四个相似性,然后将其用作疾病-微生物对的特征向量。随后,通过k-means聚类筛选出可靠的阴性样本,并进一步使用深度稀疏自编码神经网络提取疾病-微生物对的有效特征。在此基础上,提出了随机森林分类器来预测微生物与疾病之间的关联。
- 为了评估本文模型的性能,在同一数据集上实施了 10 倍交叉验证。结果,模型的 AUC 和 AUPR 分别为 0.9448 和 0.9431。此外,我们还进行了各种实验,包括阴性样本选择方法的比较、不同模型和分类器的比较、Kolmogorov-Smirnov检验和t检验、消融实验、稳健性分析以及Covid-19和结直肠癌的案例研究。结果充分证明了我们模型的可靠性和可用性。
从重叠群到染色体:长读长组装的自动改进
From contigs towards chromosomes: automatic improvement of long read assemblies (ILRA) . Brief Bioinform
- long read技术的最新进展不仅使大型联盟能够对地球上的所有真核生物进行测序,而且还允许各个实验室以相对较低的投资对其感兴趣的物种进行测序。long read技术有望克服与重复和低复杂性序列相关的支架问题,但重叠群的数量通常远远超过染色体的数量,并且它们可能在同聚物束周围包含许多插入和缺失错误。
- 为了克服这些问题,我们实施了 ILRA 管道来纠正基于long read的程序集。如果错误或污染,重叠群首先被重新排序、重命名、合并、循环或过滤。随后使用 Illumina 短读来纠正均聚物错误。
- 我们通过改进智人、布氏锥虫和细球菌属的基因组序列,并从现场样本中生成四种新型恶性疟原虫组装体,成功地测试了我们的方法。我们发现,纠正均聚物束减少了被错误注释为假基因的基因数量,但似乎需要迭代方法来纠正更多测序错误。
- 总之,我们描述了新工具的性能并对其进行了基准测试,该工具将新型长读组件的质量提高到了 1 Gbp。该管道可在 GitHub 上找到:https://github.com/ThomasDOtto/ILRA。
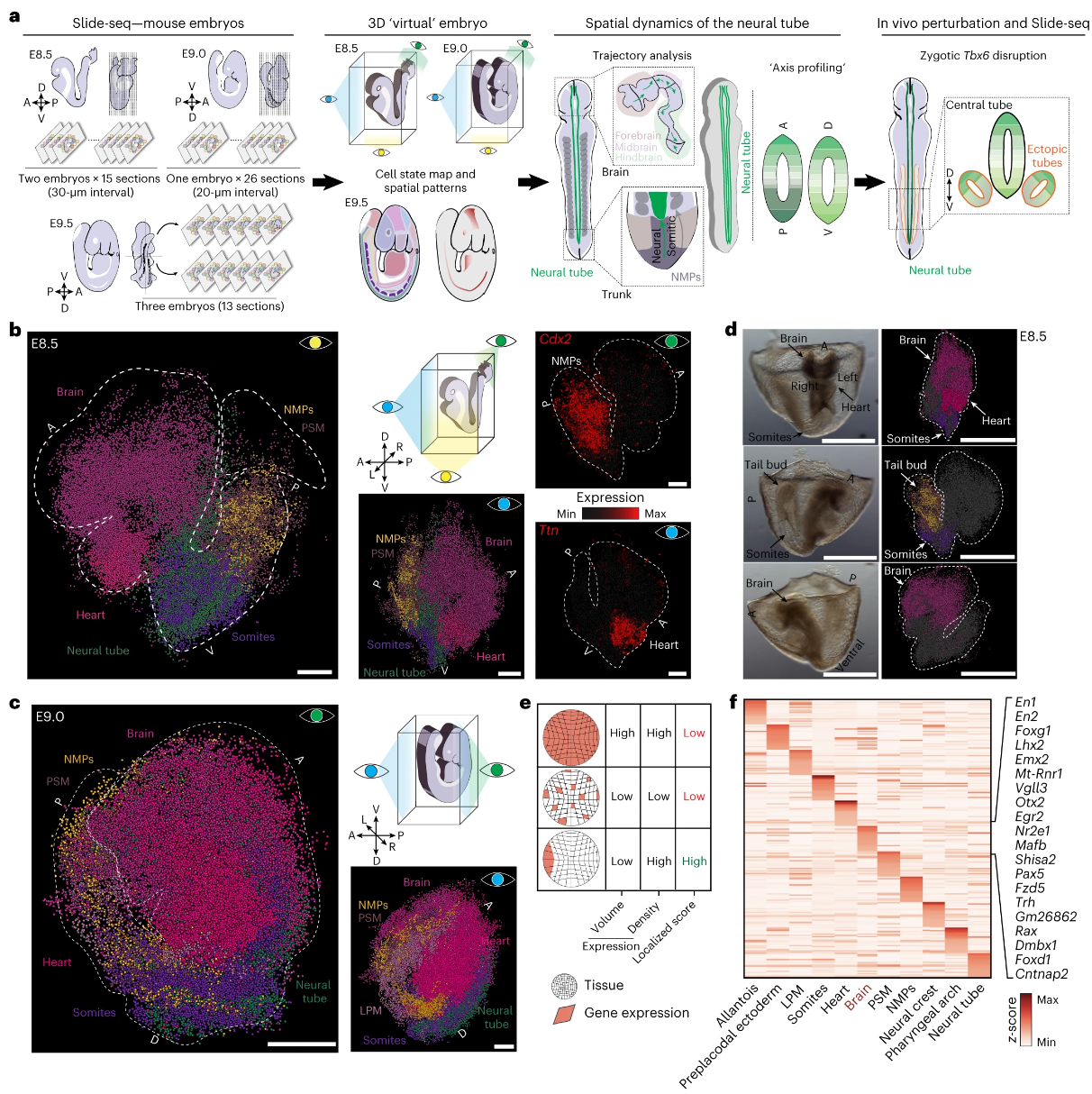
器官发生开始时全小鼠胚胎的时空转录组图
Spatiotemporal transcriptomic maps of whole mouse embryos at the onset of organogenesis. Nat Genet
- 基因表达的时空协调是胚胎正常发育所必需的。单细胞技术的使用已开始提高早期调控动力学的分辨率,包括小鼠胚胎发生过程中大多数细胞状态的详细分子定义。
- 在这里,我们使用 Slide-seq 构建完整胚胎 (E) 8.5 和 E9.0 以及部分 E9.5 胚胎的空间转录组图。
- 为了支持它们的实用性,我们开发了 sc3D,这是一种用于重建和探索三维“虚拟胚胎”的工具,可以对区域化基因表达模式进行定量研究。我们沿着发育中的神经管的主要胚胎轴进行的测量揭示了几个先前未注释的具有不同空间模式的基因。我们还描述了 Tbx6 突变胚胎中出现的“异位”神经管的相互冲突的转录特性。
- 总之,我们提出了一个用于整个胚胎结构和突变表型的时空研究的实验和计算框架。
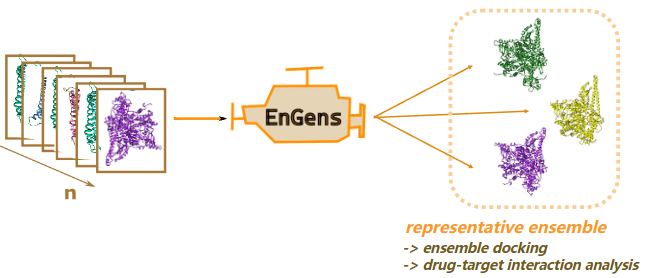
EnGens生成和分析代表性蛋白质构象集成
EnGens: a computational framework for generation and analysis of representative protein conformational ensembles. Brief Bioinform
- 蛋白质是在细胞中执行重要功能的动态大分子。蛋白质的结构决定了其功能,但这种结构不是静态的,因为蛋白质会改变其构象以实现各种功能。了解蛋白质的构象景观对于了解其作用机制至关重要。精心选择的构象集可以概括这种复杂的景观,并比单一构象提供更好的蛋白质功能见解。我们将这些集合称为代表性构象集合。计算方法的最新进展导致跨越构象景观的可用结构数据集数量的增加。然而,从此类数据集中提取代表性构象集合并不是一件容易的任务,并且已经开发了许多方法来解决它。
- 我们的新方法 EnGens(集成生成的缩写)将这些方法收集到一个统一的框架中,用于生成和分析代表性蛋白质构象集成。在这项工作中,我们:(1)概述了用于代表性蛋白质结构整体生成和分析的现有方法和工具; (2) 将现有方法统一到开源 Python 包和可移植 Docker 映像中,在 Jupyter Notebook 管道中提供交互式可视化; (3) 使用文献中的一些典型示例来测试我们的流程。
- EnGens 产生的代表性集成体可用于许多下游任务,例如蛋白质-配体集成体对接、蛋白质动力学的马尔可夫状态建模以及单点突变效应分析。
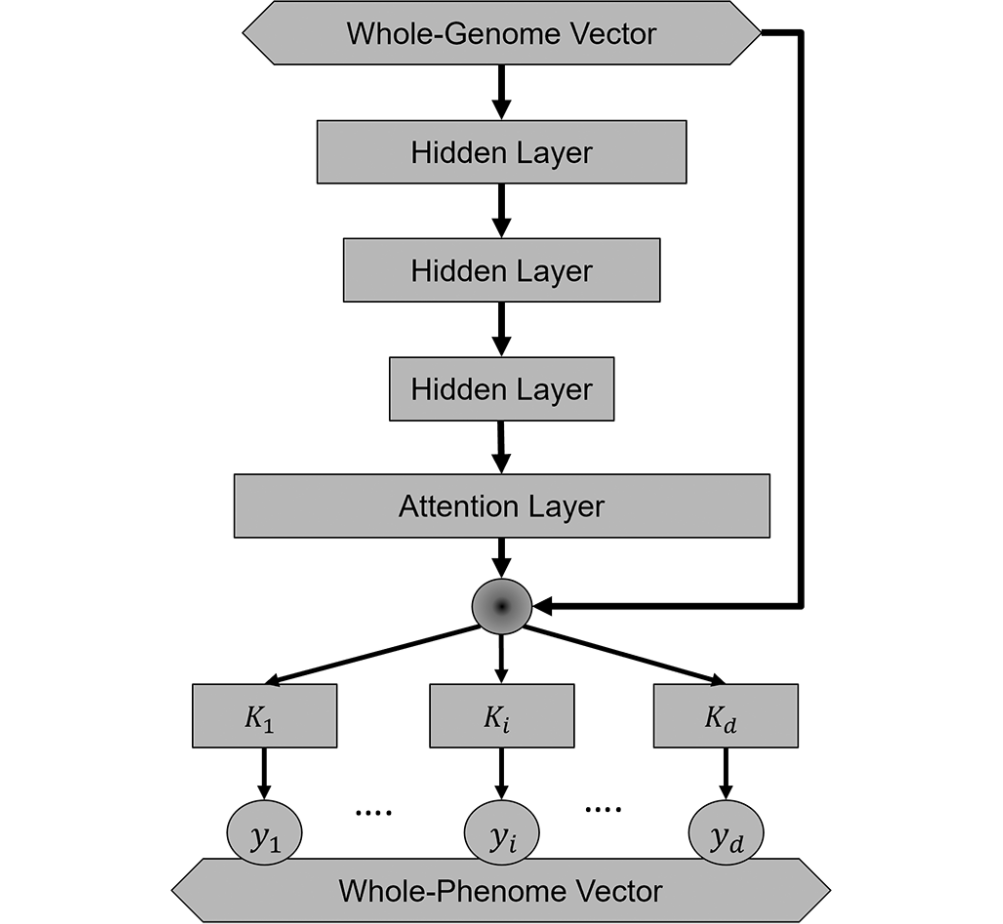
(~ ̄▽ ̄)~ 多任务学习改进泛癌多基因风险评分的性能
Explainable multi-task learning improves the parallel estimation of polygenic risk scores for many diseases through shared genetic basis. PLoS Comput Biol
了解一下作者测试泛癌模型的具体策略
对GSClassifier可能有帮助
- 许多复杂疾病具有共同的遗传决定因素,并且在人群中共病。我们假设,可以利用疾病的共存及其重叠的遗传病因来同时改善多种疾病的多基因风险评分(PRS)。使用基于可解释的神经网络架构的多任务学习(MTL)方法测试了这一假设。
- 我们发现,在泛癌症 MTL 模型中对 17 种常见癌症的 PRS 进行并行估计通常比在可比较的单任务学习 (STL) 模型中对单个癌症进行独立估计更准确。在泛疾病 MTL 模型中,对于 60 种流行的非癌症疾病,也一致观察到积极迁移学习带来的这种性能改进。
- MTL 模型的解释揭示了神经网络用于 PRS 估计的重要单核苷酸多态性组之间存在显着的遗传相关性。这表明具有共同遗传基础的紧密联系的疾病网络。
序列划线草图与比较系统
Genomic sketching with multiplicities and locality-sensitive hashing using Dashing 2. Genome Res
- 基因组草图是测序数据集中 k 聚体集的小型概率表示。草图是大规模分析的构建块,考虑了多对序列或序列集合之间的相似性。虽然现有工具可以轻松比较数以万计的基因组,但相关数据集可以达到数百万个序列甚至更多。流行的工具也未能考虑 k 聚体的多重性,这使得它们不太适用于定量设置。
- 我们描述了一种名为 Dashing 2 的方法,该方法构建在 SetSketch 数据结构上。 SetSketch 与 HyperLogLog 相关,但放弃使用前导零计数,转而使用可调整基数的截断对数。与 HLL 不同,SetSketch 与 ProbMinHash 方法结合使用时可以执行多重性感知草图。 Dashing 2 集成了局部敏感哈希,可将所有对的比较扩展到数百万个序列。
- Dashing 2 是免费的开源软件。
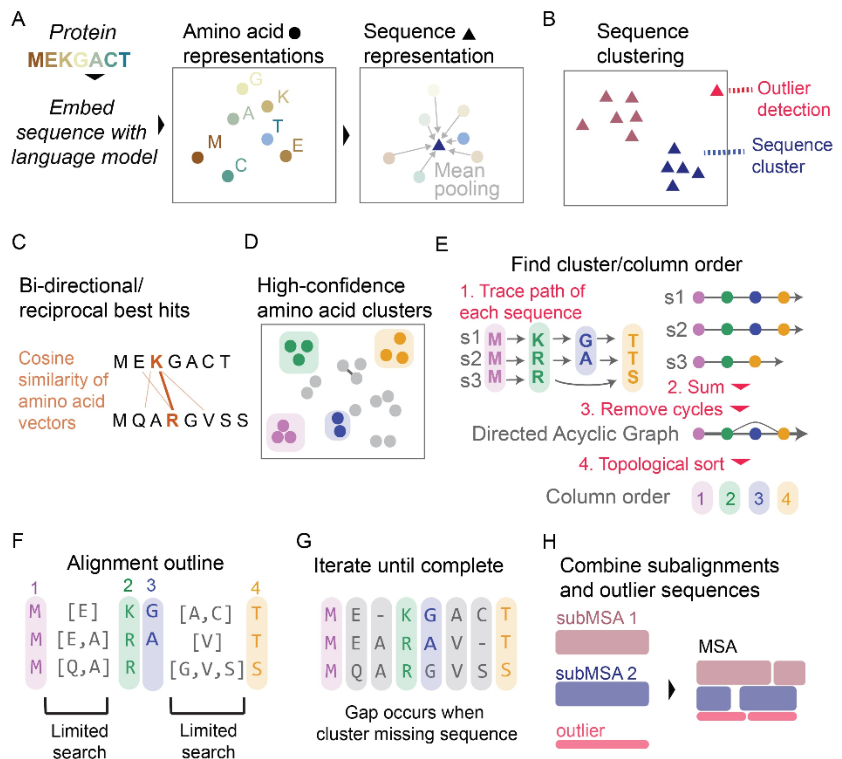
利用蛋白质语言模型进行准确的多序列比对
Leveraging protein language models for accurate multiple sequence alignments. Genome Res
- 多序列比对(Multiple sequence alignment, MSA)是蛋白质序列和功能研究的关键步骤。通常,多个序列比对算法逐步比对序列对,并在引导树的帮助下组合这些比对。这些比对算法使用基于替换矩阵的评分系统来测量氨基酸相似性。尽管标准方法取得了成功,但在处理序列同一性较低的蛋白质组(即所谓的蛋白质比对暮光区)时却遇到了困难。对于这些困难的情况,需要另一个信息来源。蛋白质语言模型是一种强大的新方法,它利用大量序列数据集为序列中的每个氨基酸生成高维上下文嵌入。这些嵌入已被证明反映了蛋白质内氨基酸的物理化学和高阶结构和功能属性。
- 在这里,我们提出了一种基于聚类和排序氨基酸上下文嵌入的多序列比对新方法。我们用于比对语义一致的蛋白质组的方法避免了对多序列比对算法的许多标准组件的需要,避免了初始引导树构建、中间成对比对、间隙罚分和替换矩阵。来自上下文嵌入的附加信息可以提高结构相似且氨基酸相似度较低的蛋白质的比对精度。
- 我们预计蛋白质语言模型将成为下一代 MSA 生成算法的基本组成部分。
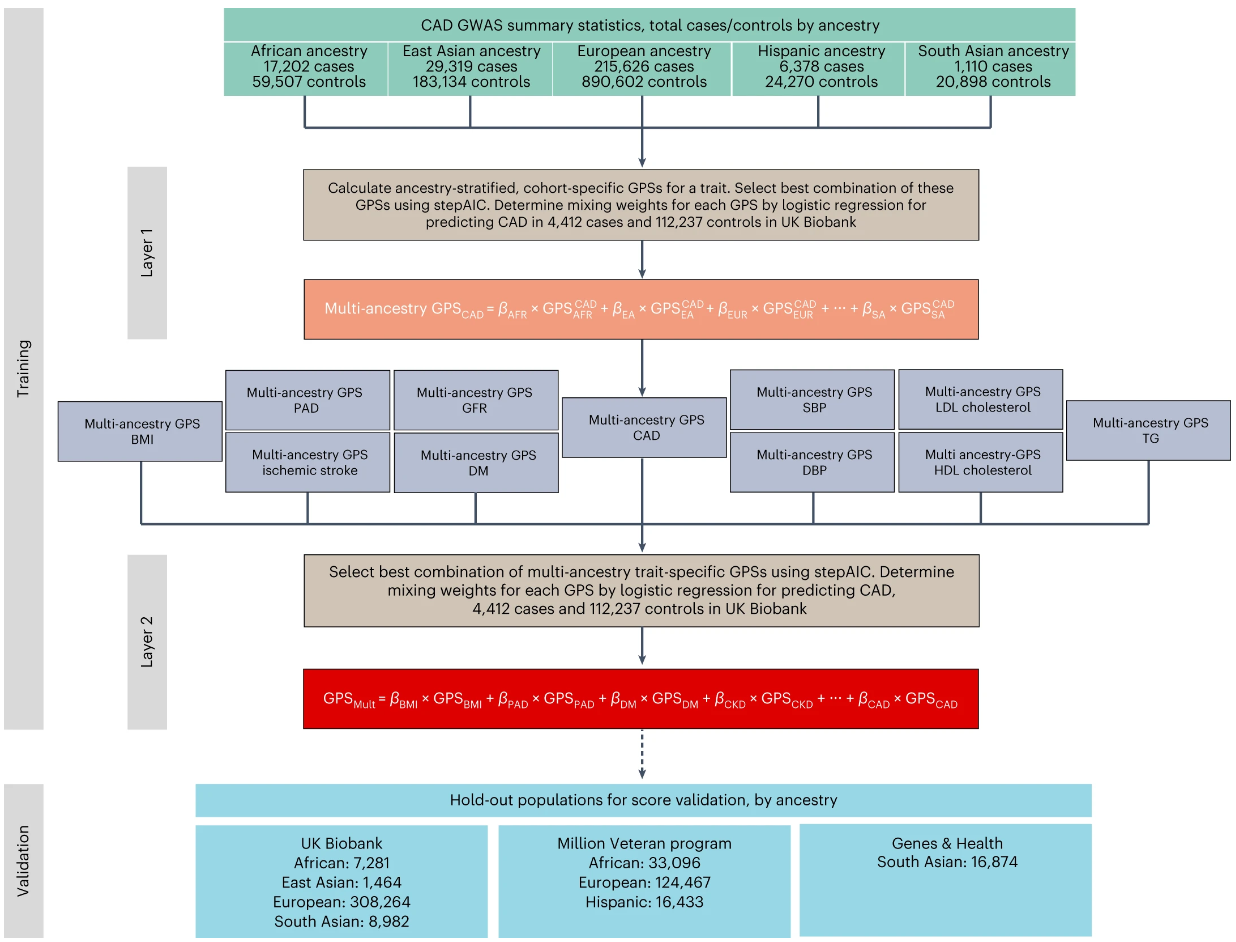
冠状动脉疾病的多基因风险评分系统GPSMult
A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat Med
- 识别冠状动脉疾病 (CAD) 风险最高的个体(最好是在发病前)仍然是一项重要的公共卫生需求。先前的研究已经开发出全基因组多基因评分来实现风险分层,反映了 CAD 风险的重要遗传因素。
- 在这里,我们开发了一种新的、显着改进的 CAD 多基因评分,称为 GPSMult,它整合了 5 个 CAD 祖先(>269,000 例病例和 >1,178,000 例对照)的全基因组关联数据和 10 个 CAD 风险因素。
- GPSMult 与欧洲血统的英国生物银行参与者中流行的 CAD 密切相关(每标准差的比值比为 2.14,95% 置信区间为 2.10-2.19,P < 0.001),确定 20.0% 的人群风险增加 3 倍,反之则为 13.9%与中间五分之一的人相比,风险降低了 3 倍。 GPSMult 还与 CAD 事件相关(每标准差的风险比为 1.73,95% 置信区间为 1.70-1.76,P < 0.001),识别出 3% 的健康个体未来发生 CAD 事件的风险与患有现有疾病的人相当,并且显着改善风险歧视和重新分类。
- 在分别包含 33,096、124,467、16,433 和 16,874 名非洲裔、欧洲裔、西班牙裔和南亚裔参与者的多种族外部验证数据集中,GPSMult 证明了所有血统之间的关联强度有所增加,并且优于所有先前发布的 CAD 多基因评分。
- 这些数据为该领域贡献了新的 GPSMult for CAD,并提供了一个通用框架,说明大规模整合 CAD 遗传关联数据和来自不同人群的相关性状如何能够有意义地改进多基因风险预测。
高通量单细胞分析数据集中体细胞突变的从头检测
De novo detection of somatic mutations in high-throughput single-cell profiling data sets. Nat Biotechnol
英国欧洲生物信息学研究所欧洲分子生物学实验室
很好的研究,多多关注
- 单细胞分辨率的体细胞突变表征对于研究癌症进化、克隆镶嵌和细胞可塑性至关重要。
- 在这里,我们描述了 SComatic,一种设计用于直接检测单细胞转录组和 ATAC-seq(转座酶可访问染色质序列的分析)数据集中的体细胞突变的算法,无需匹配的批量或单细胞 DNA 测序数据。 SComatic 使用过滤器和非肿瘤样本参数化的统计测试将体细胞突变与多态性、RNA 编辑事件和假象区分开来。
- 使用来自涵盖癌症和非肿瘤样本的 688 个单细胞 RNA-seq (scRNA-seq) 和单细胞 ATAC-seq (scATAC-seq) 数据集中的超过 260 万个单细胞,我们表明 SComatic 可以检测单细胞中的突变准确地,即使是来自多克隆组织的分化细胞,这些细胞不适合使用现有方法进行突变检测。根据匹配的基因组测序和 scRNA-seq 数据进行验证,SComatic 在不同数据集上的 F1 得分在 0.6 到 0.7 之间,而表现第二好的方法的 F1 得分为 0.2-0.4。
- 总之,SComatic 允许从头突变特征分析,以及单细胞分辨率的克隆异质性和突变负担研究。
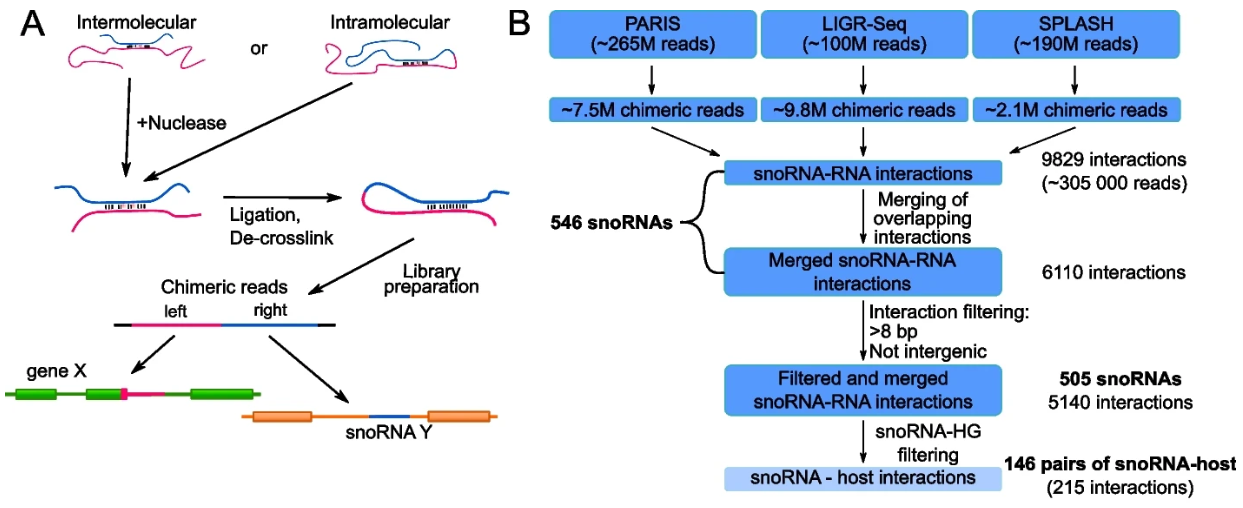
内含子snoRNA与宿主转录本的相互作用很普遍
Intronic small nucleolar RNAs regulate host gene splicing through base pairing with their adjacent intronic sequences. Genome Biol
- 小核仁 RNA (snoRNA) 是丰富的非编码 RNA,因其参与核糖体 RNA 成熟而闻名。在哺乳动物中,大多数表达的 snoRNA 嵌入较长基因的内含子中,并通过宿主的转录和剪接产生。内含子 snoRNA 长期以来被视为惰性乘客,对宿主表达影响很小。然而,最近的一项研究报道了 snoRNA 影响其宿主基因的剪接和最终输出。总体而言,内含子 snoRNA 对宿主表达的一般贡献仍不清楚。
- 大规模人类 RNA-RNA 相互作用数据集的计算分析表明,检测到的 snoRNA 中有 30% 与其宿主转录本相互作用。许多 snoRNA 宿主双链体位于选择性剪接的外显子附近,并表现出高度的序列保守性,表明在剪接调节中可能发挥作用。
- 对 SNORD2-EIF4A2 双链体模型的研究表明,snoRNA 与宿主内含子序列的相互作用隐藏了分支点,导致相邻替代外显子的包含减少。包含相互作用的内含子区域的扩展 SNORD2 序列以细胞类型特异性的方式在测序数据集中累积。反义寡核苷酸和破坏 snoRNA 内含子结构形成的突变促进替代外显子的剪接,使 EIF4A2 转录物比率远离无义介导的衰减。
- 总之,许多 snoRNA 在其宿主转录本的替代外显子附近形成 RNA 双链体,将它们置于控制宿主输出的最佳位置,如 SNORD2-EIF4A2 模型系统所示。总的来说,我们的研究支持内含子 snoRNA 在调节宿主转录成熟中发挥更广泛的作用。
(~ ̄▽ ̄)~ 变异效应图谱:以核苷酸分辨率了解基因组
An Atlas of Variant Effects to understand the genome at nucleotide resolution. Genome Biol
这个问题确实很重要
- 测序已经揭示了数亿个人类基因变异,继续努力只会加剧这种变异雪崩。目前没有足够的信息来解释大多数变异的影响,限制了精准医学和理解基因组功能的机会。
- 解决方案在于对变异的功能效果进行实验评估,这可以揭示它们的生物学和临床影响。然而,变异效应测定通常仅在首次观察之后(在大多数情况下是在其第一次观察之后很久)才对个体变异进行反应性分析。现在,变异效应的多重分析可以同时表征大量变异,产生变异效应图,揭示基因或调控元件中每个可能的单核苷酸变化的功能。
- 为人类基因组中的每个蛋白质编码基因和调控元件生成图谱将创建变异效应图谱的“图谱”,并改变我们对遗传学的理解,并开创基因组核苷酸分辨率功能知识的新时代。图谱将揭示人类基因组的基本生物学,为人类进化提供信息,促进治疗方法的开发和使用,并最大限度地利用基因组学来诊断和治疗疾病。
- 变异效应联盟图谱是一个由数百名研究人员、技术人员和临床医生组成的国际合作小组,致力于实现变异效应图谱,以帮助实现基因组学的承诺。
长寿哺乳动物的基因组比较
中国科学院动物生态与保护生物学重点实验室
- 裸鼹鼠(Heterocephalus glaber)、蝙蝠(例如鼠耳蝠属)和大象(象科)被称为长寿哺乳动物,并被认为是极好的癌症拮抗剂。然而,这些长寿物种是否存在支持癌症抵抗力的共同基因变化尚未完全确定。
- 在这里,我们新生成了高质量的染色体水平亚洲象(Elephas maximus)基因组,并确定大象中扩展的基因家族参与 Ras 相关和碱基切除修复途径。
- 此外,我们对 12 种哺乳动物进行了比较基因组分析,并检查了大象、裸鼹鼠和大马蹄蝠中具有正选择特征的基因。与短命哺乳动物相比,长寿哺乳动物中 CDR2L 和 ALDH6A1 阳性选择位点的残基增强了对肿瘤细胞迁移的抑制作用。
- 总的来说,我们的研究提供了一种新的基因组资源,并对长寿哺乳动物的常见遗传变化进行了初步调查。
mapquik:最小化空间中长读取的高效低发散映射
Efficient mapping of accurate long reads in minimizer space with mapquik. Genome Res
麻省理工学院
- DNA 测序数据不断向更长的读取方向发展,并且测序错误率越来越低。我们专注于将长读长(例如 PacBio HiFi)的低分歧序列映射或比对到参考基因组的关键问题,这在使用尖端读长映射方法时在准确性和计算资源方面提出了挑战专为所有类型的对齐而设计。一个自然的想法是使用更长的种子来优化效率,以减少无关匹配的可能性;然而,连续的精确种子很快就会达到灵敏度极限。
- 我们引入了mapquik,一种新颖的策略,通过k个连续采样的最小化器(k-min-mers)的匹配来锚定比对,并仅索引在参考基因组中出现一次的k-min-mers,从而创建准确的更长种子,从而解锁超长种子。快速测绘,同时保持高灵敏度。
- 我们证明,mapquik 显着加速了人类和玉米基因组的播种和链接步骤(读取图谱的基本瓶颈),具有 > 96% 的灵敏度和近乎完美的特异性。在人类基因组上,无论是真实读取还是模拟读取,mapquik 都比最先进的工具 minimap2 实现了 37 倍的加速,在玉米基因组上,比 minimap2 实现了 410 倍的加速,这使得 Mapquik 成为迄今为止最快的地图绘制器。
- 这些加速不仅可以通过最小化空间播种实现,还可以通过一种新颖的启发式 O(n) 伪链接算法实现,该算法改进了长期存在的 O(n log n) 界限。最小化空间计算为实现长读长测序数据的实时分析奠定了基础。
- 代码:ekimb/mapquik: Efficient low-divergence mapping of long reads in minimizer space
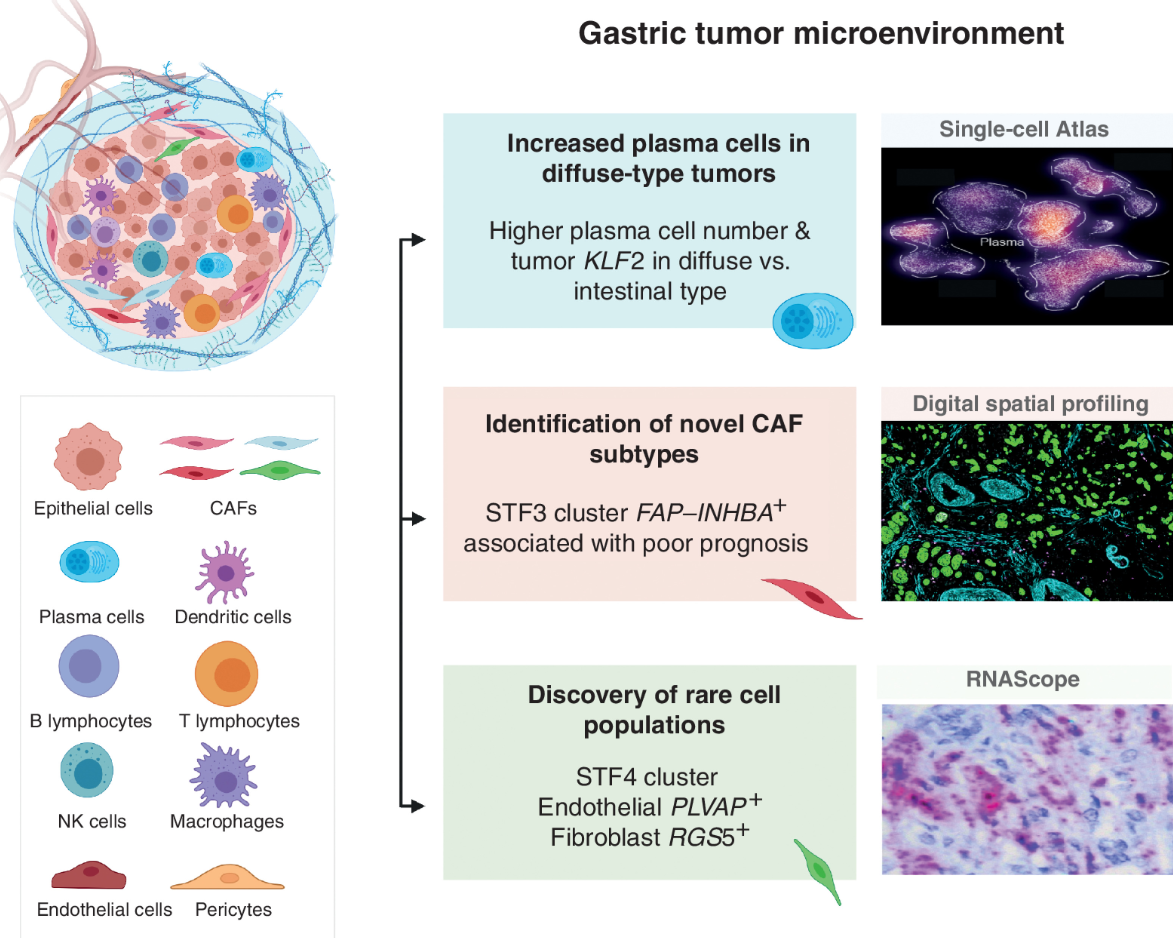
(~ ̄▽ ̄)~ 2022-胃癌单细胞组学分析
Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. full html. Data:GSE183904
- 胃癌异质性是疾病管理的障碍。我们生成了一份全面的胃癌单细胞图谱(>200,000 个细胞),其中包含来自 31 名患者的 48 个样本,涵盖临床分期和组织学亚型。
- 我们鉴定了 34 种不同的细胞谱系状态,包括新型稀有细胞群。许多谱系状态表现出独特的癌症相关表达谱,分别促成肿瘤范围内的分子拼贴。我们观察到与上皮驻留 KLF2 相关的弥漫型肿瘤中浆细胞比例增加,以及以高 INHBA 和 FAP 共表达为标志的癌症相关成纤维细胞亚群的阶段性增长。
- 患者来源的类器官 (PDO) 和原发性肿瘤之间的单细胞比较突出了谱系间和谱系内的相似性和差异,划分了作为实验模型的 PDO 的分子边界。我们通过空间转录组学、独立批量 RNA 测序队列的正交验证以及使用体外和体内模型的功能演示来补充这些发现。
- 我们的结果提供了不同胃癌亚型的患者内和患者间谱系状态的高分辨率分子资源。
人类基因组版本T2T-CHM13 vs. GRCh38的差异
Characterization of large-scale genomic differences in the first complete human genome. Genome BIol
上海交通大学-发育与神经精神疾病遗传学教育部重点实验室-Bio-X 研究所
- 首个端粒到端粒(T2T)人类基因组组装(T2T-CHM13)的发布是人类基因组学的里程碑。 T2T-CHM13 基因组组装扩展了我们对端粒、着丝粒、片段复制和其他复杂区域的理解。目前人类基因组参考(GRCh38)已广泛应用于各种人类基因组研究。然而,这两个重要基因组组装之间的大规模基因组差异尚未得到详细表征。
- 在这里,除了之前报道的“非同线性”区域之外,我们还发现了 67 个额外的大规模差异区域,并使用新开发的名为 SynPlotter 的网站工具将它们精确地分为四种结构类型。不包括端粒和着丝粒区域的差异区域(~ 21.6 Mbp)在人类中具有高度结构多态性,其中缺失或重复可能与各种人类疾病有关,例如免疫和神经发育障碍。对新发现的差异区域(KLRC 基因簇)的分析表明,单缺失事件导致的 KLRC2 缺失与约 20% 的人类自然杀伤细胞分化相关。同时,KLRC3 中观察到的快速氨基酸替换可能是灵长类动物进化中自然选择的结果。
- 我们的研究为理解两个关键人类参考基因组之间的大规模结构基因组差异奠定了基础,因此对未来的人类基因组学研究非常重要。
综述类
肠道微生物与肿瘤
Bacteria in cancer initiation, promotion and progression. Nat Rev Cancer
- 癌细胞起源于一系列获得性基因突变,这些突变可以驱动其不受控制的细胞增殖和免疫逃避。环境因素,包括寄生在人体的微生物,可以改变肿瘤细胞的新陈代谢、生长模式和功能,并塑造肿瘤微环境。肠道微生物群失调现在被科学界认为是癌症的标志。
- 然而,只有少数微生物被发现可以直接引发肿瘤发生或扭曲免疫系统以产生肿瘤生长环境。在过去的二十年中,对人类微生物组及其在个体内部和个体之间的功能的研究揭示了以微生物群为中心的健康和疾病策略。
- 在这里,我们回顾了对微生物群在癌症发生、促进和进展中作用机制的不断发展的理解。我们探索细菌在胃肠道恶性肿瘤以及肺癌、乳腺癌和前列腺癌中的作用。最后,我们讨论了在个性化癌症预防、诊断和治疗中靶向或利用细菌的前景和局限性。
mRNA:癌症免疫治疗的一个有前途的平台
mRNA: A promising platform for cancer immunotherapy. Adv Drug Deliv Rev
- 由于在 COVID-19 大流行期间使用脂质纳米颗粒技术的 mRNA 疫苗取得了显着的临床效果,信使 RNA (mRNA) 作为治疗各种人类疾病,特别是恶性肿瘤的强大工具而受到关注。
- 最近有希望的临床前和临床结果集中体现了 mRNA 和基于纳米制剂的递送技术的进步,凸显了 mRNA 在癌症免疫治疗中的巨大潜力。 mRNA 可以以各种治疗方式用于癌症免疫治疗,包括癌症疫苗、过继性 T 细胞疗法、治疗性抗体和免疫调节蛋白。
- 这篇综述全面概述了基于 mRNA 的疗法的现状和前景,包括众多的递送和治疗策略。
人类癌症中肿瘤浸润 B 细胞的生发中心依赖性和非依赖性免疫反应
Germinal center-dependent and -independent immune responses of tumor-infiltrating B cells in human cancers. Cell Mol Immunol
- B 细胞在免疫中发挥重要作用,主要通过产生高亲和力浆细胞 (PC) 和记忆 B (Bmem) 细胞。 B 细胞的亲和力成熟和分化分别依赖于抗原结合和微环境提供的 B 细胞受体 (BCR) 内在和外在信号的整合。近年来,肿瘤浸润 B (TIL-B) 细胞和 PC (TIL-PC) 已被发现是人类癌症抗肿瘤反应的重要参与者,但它们的相互作用和动态仍然很大程度上未知。
- 在淋巴器官中,B 细胞反应涉及 Bmem 细胞和 PC 产生的生发中心 (GC) 依赖性和 GC 独立途径。 BCR 库的亲和力成熟发生在具有 B 细胞信号整合的特定时空动态的 GC 反应中。一般来说,抗原对高亲和力 Bmem 细胞的重新激活会触发不依赖 GC 的大量 PC 的产生,而无需 BCR 再多样化。了解免疫反应中的 B 细胞动态需要整合多种工具和读数,例如单细胞表型分析和 RNA-seq、原位分析、BCR 谱分析、BCR 特异性和亲和力测定以及功能测试。
- 在这里,我们回顾了最近如何应用这些工具来研究不同类型实体瘤中的 TIL-B 细胞和 TIL-PC。我们评估了已发表的不同 TIL-B 细胞动力学模型的证据,涉及 GC 依赖性或 GC 独立的局部反应以及由此产生的抗原特异性 PC。
- 总而言之,我们强调需要进行更综合的 B 细胞免疫学研究,以合理地研究 TIL-B 细胞作为抗肿瘤治疗的杠杆。
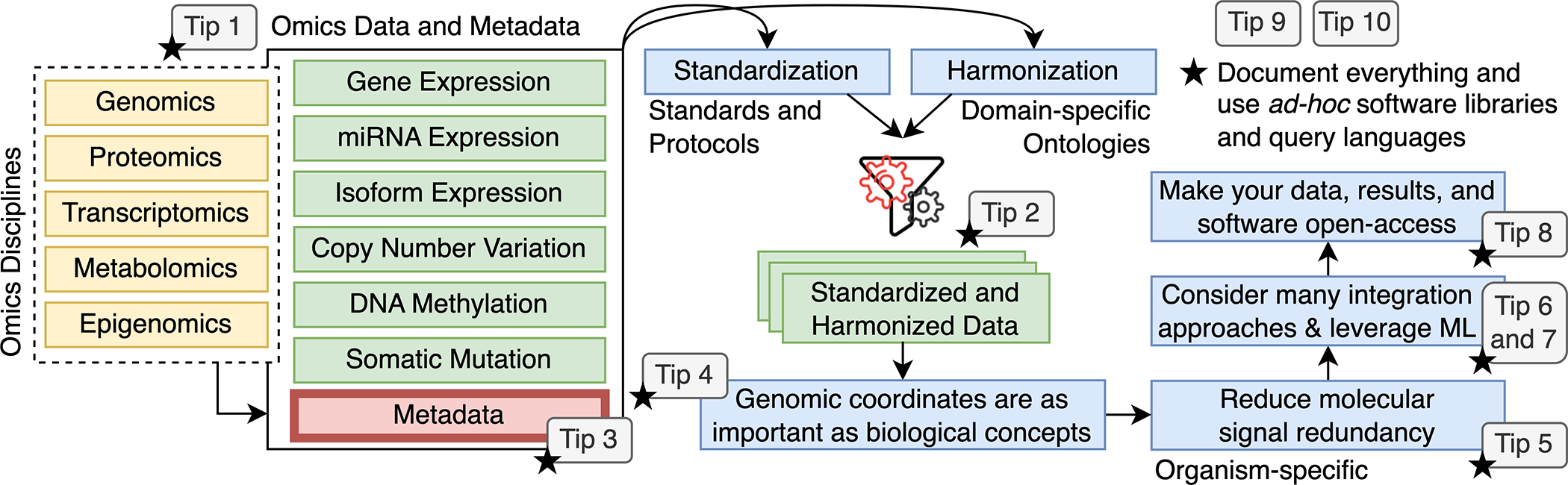
避免多组学数据集成分析中的陷阱的十个快速技巧
Ten quick tips for avoiding pitfalls in multi-omics data integration analyses. PLoS Comput Biol
- 数据是生物信息学最重要的元素:事实上,生物信息学数据的计算分析可以帮助研究人员推断有关生物学、化学、生物物理学,有时甚至是医学的新知识,影响对患者的治疗和治疗。来自不同来源的生物信息学和高通量生物数据甚至可能更有帮助,因为每个不同的数据块都可以提供有关特定生物现象的替代、补充信息,类似于从不同角度拍摄同一主题的多张照片。
- 在这种背景下,生物信息学和高通量生物数据的整合在成功进行生物信息学研究中发挥着关键作用。在过去的几十年中,源自蛋白质组学、代谢组学、宏基因组学、表型组学、转录组学和表观基因组学的数据已被标记为组学数据,作为引用它们的唯一名称,并且这些组学数据的整合在所有生物领域中变得越来越重要。即使这种组学数据集成有用且相关,但由于其异质性,在集成阶段出错的情况并不少见。
- 因此,我们决定提出这十个快速提示,以正确执行组学数据集成,避免我们在过去发表的研究中经历或注意到的常见错误。即使我们通过使用(我们希望)任何人都可以理解的简单语言为初学者设计了十项指南,我们相信所有进行组学数据集成的生物信息学家(包括专家)都应该考虑我们的十项建议。
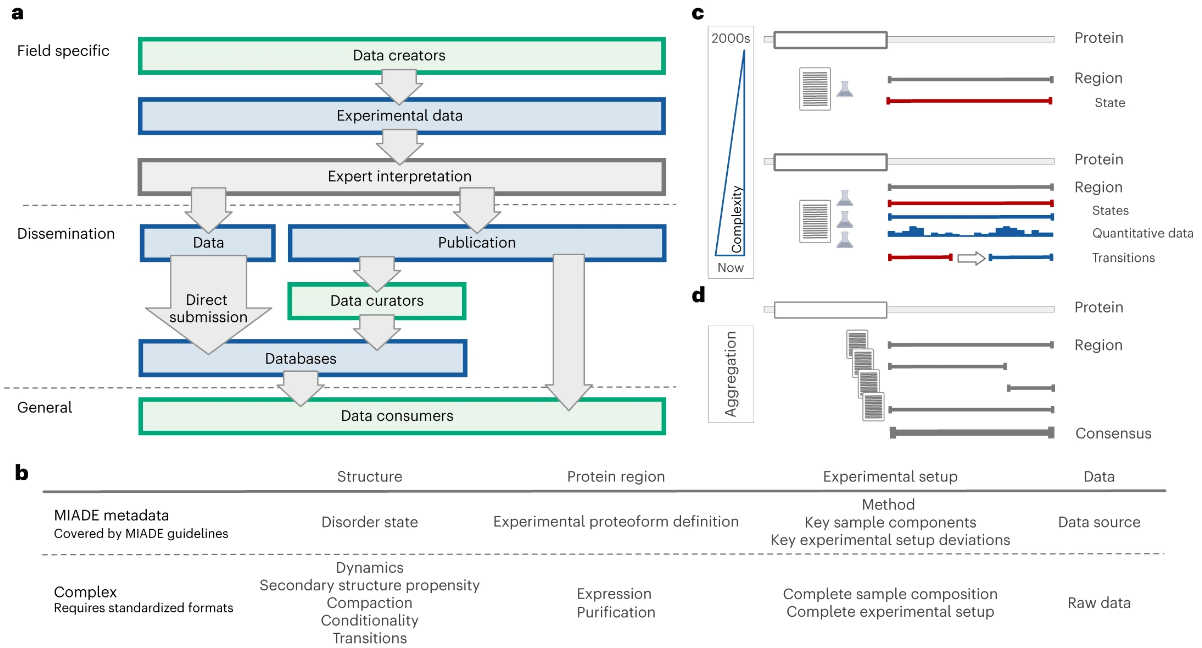
本质无序蛋白的研究指南
Minimum information guidelines for experiments structurally characterizing intrinsically disordered protein regions. Nat Methods
- 对实验以及随后的生物学观察的明确描述对于准确的数据解释至关重要。最低信息指南定义了数据的基本补充,可以支持基于实验观察的明确结论。
- 我们提出了有关无序实验的最低信息 (Minimum Information About Disorder Experiments, MIADE) 指南,以定义更广泛的科学界所需的参数,以了解研究本质无序区域 (IDR) 结构特性的实验结果。 MIADE 指南为数据生产者提供了从源头描述实验结果的建议,为管理者向社区资源注释实验数据以及为维护社区资源的数据库开发人员提供了传播数据的建议。 MIADE 指南将提高数据消费者实验结果的可解释性,促进直接数据提交,简化数据管理,改善存储库之间的数据交换,并标准化 IDR 数据源对 IDR 实验关键元数据的传播。
流行病学类
- 暂无。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍

看到华为的什么盘古,估计就得烂尾
学术上的成功和商业上的成功是不同的。看前沿主要是为了了解研究现状(或者说,审稿人的taste),也不是想要follow某个团队的工作。能发表在Nature的研究,虽然不见得一定是什么经典之作,但一般都有独到之处,学习一下并无大碍 (ฅ´ω`ฅ)