本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- 组装质量控制中的孤儿亚基识别机制
- 孤儿质量控制塑造网络动态和基因表达
- 超快远距离伤口反应对于全身再生至关重要
- 六体-INO80复合物揭示了非典型核小体重塑的结构…
- INO80 在六体上的重新定位揭示了机械多功能性的…
前言
本文是前沿快讯的第35期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。从第18期开始,“肿瘤转移类”、“肿瘤代谢类”等将不再更新,而是合并至其它分类。
CNS类
组装质量控制中的孤儿亚基识别机制
Mechanism of orphan subunit recognition during assembly quality control. Cell. full html
系列研究
英国MRC 分子生物学实验室
subunit是一个蛮有趣的概念,可以多多了解
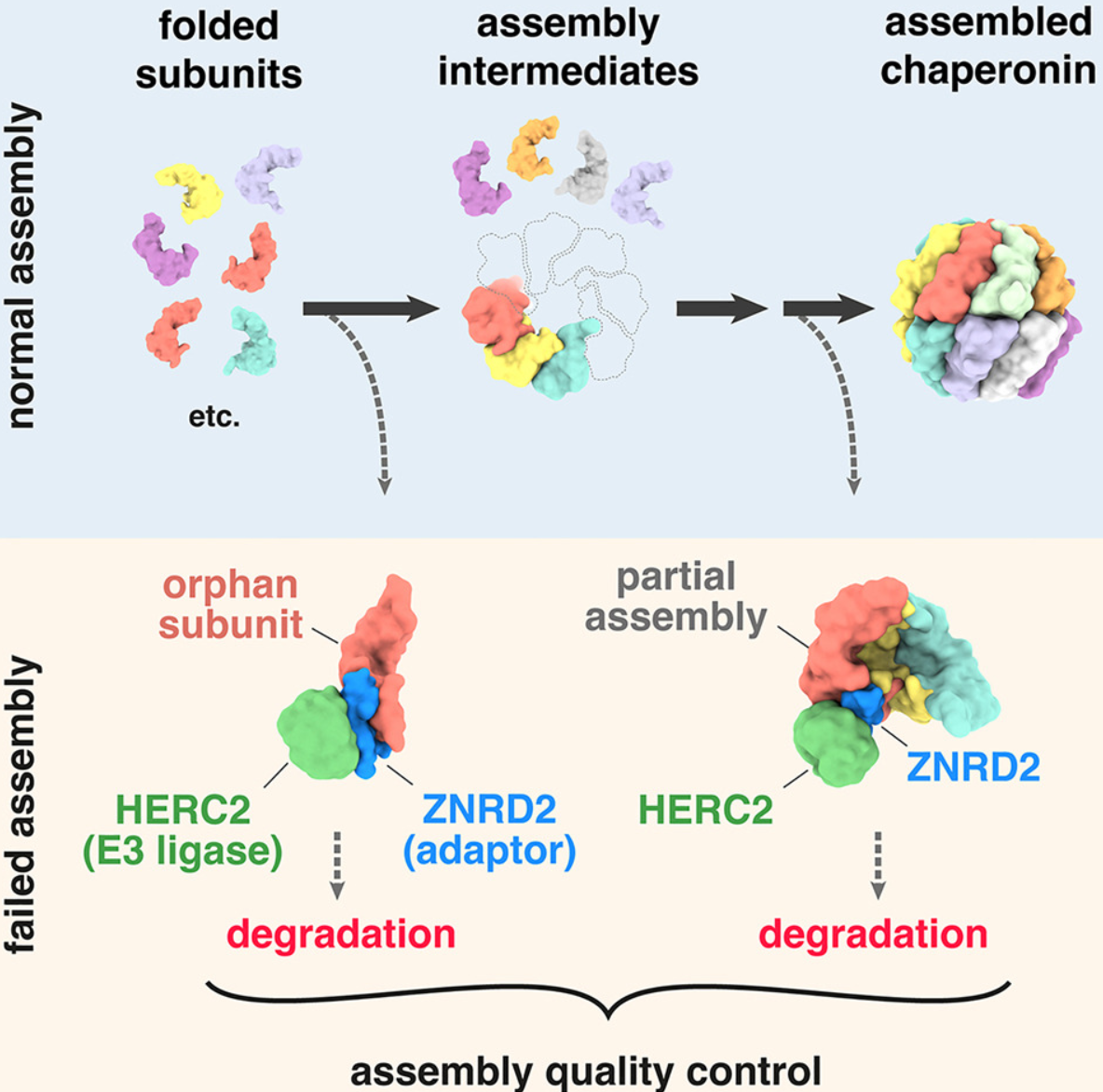
- 细胞含有许多由多个亚基组装而成的丰富的分子机器。亚基生产的不平衡和组装失败会产生孤儿亚基,这些孤儿亚基会被不明确的途径消除。
- 在这里,我们确定了胞质伴侣蛋白 CCT 的孤儿亚基是如何被识别的。几个未组装的 CCT 亚基使用 ZNRD2 作为接头来招募 E3 泛素连接酶 HERC2。这两个因子对于细胞中孤儿 CCT 亚基降解是必需的,对于用纯化因子进行 CCT 亚基泛素化来说足够,并且是最佳细胞适应性所必需的。
- 域映射和结构预测定义了最小 HERC2-ZNRD2-CCT 模块的分子特征。该结构模型的关键要素在细胞中使用点突变体进行了验证,表明了为什么 ZNRD2 选择性地识别多个孤立的 CCT 亚基而不参与组装的 CCT。
- 我们的研究结果揭示了如何监控 CCT 组装过程中的失败,并为孤儿亚基的分子识别提供了范例,孤儿亚基是细胞中质量控制底物的最大来源。
孤儿质量控制塑造网络动态和基因表达
Orphan quality control shapes network dynamics and gene expression. Cell
系列研究
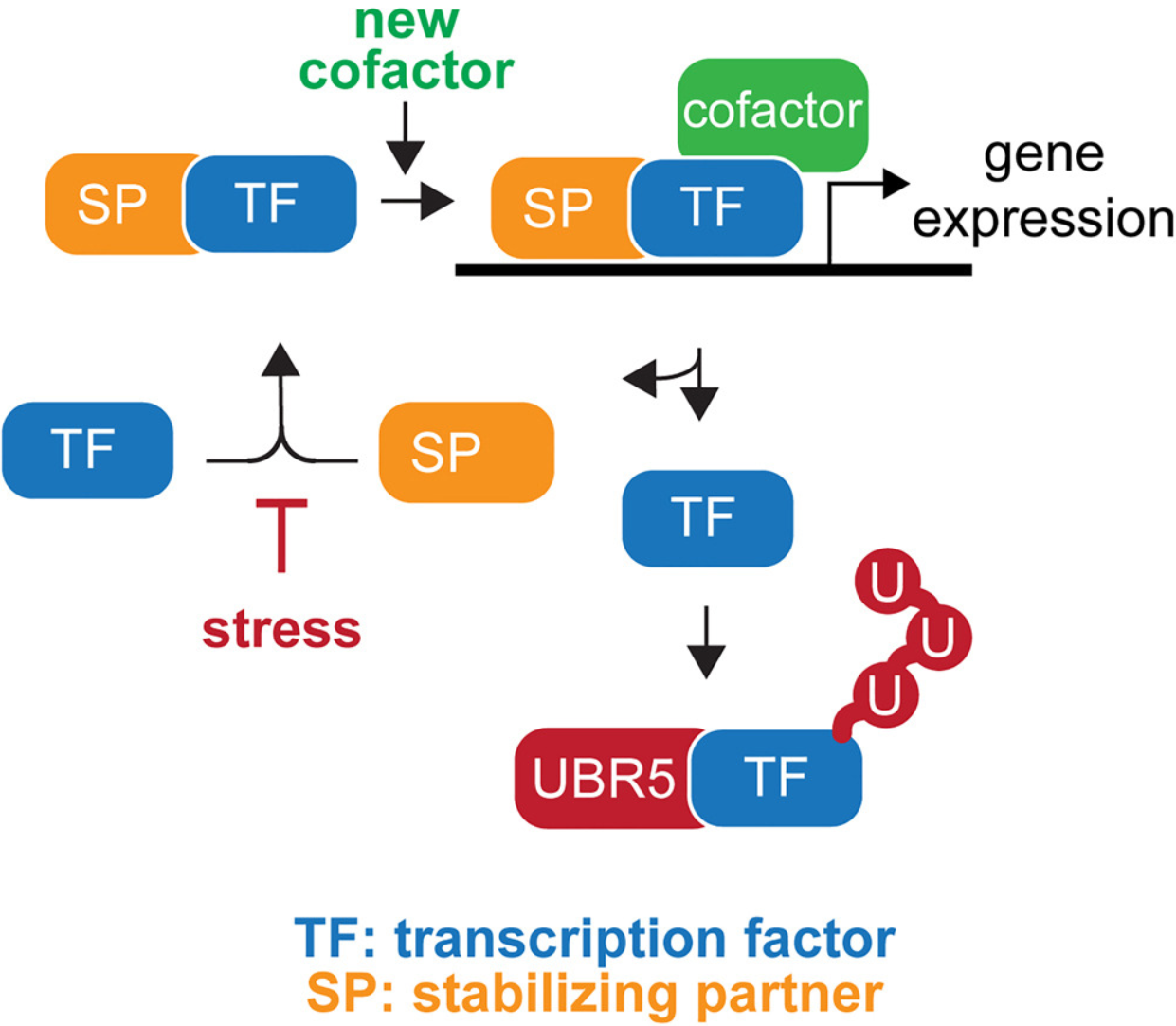
- 所有真核生物都需要复杂的蛋白质网络来将发育信号转化为准确的细胞命运决定。扰乱网络组件之间相互作用的突变通常会导致疾病,但复杂网络的组成和动态是如何建立的仍然知之甚少。
- 在这里,我们将 E3 连接酶 UBR5 确定为信号中枢,有助于降解在以 c-Myc 癌蛋白为中心的网络中起作用的多个转录调节因子的不配对亚基。生化和结构分析表明,UBR5 结合的基序只有在复杂解离后才可用。通过快速翻转未配对的转录因子亚基,UBR5 在转录调节因子之间建立动态相互作用,使细胞能够有效地执行基因表达,同时保持对环境信号的接受。
- 我们得出的结论是,孤儿质量控制在建立动态蛋白质网络中起着至关重要的作用,这可能解释了转录过程中蛋白质降解的保守需求,并为调节疾病中的基因表达提供了机会。
超快远距离伤口反应对于全身再生至关重要
Ultrafast distant wound response is essential for whole-body regeneration. Cell
美国斯坦福大学生物工程系
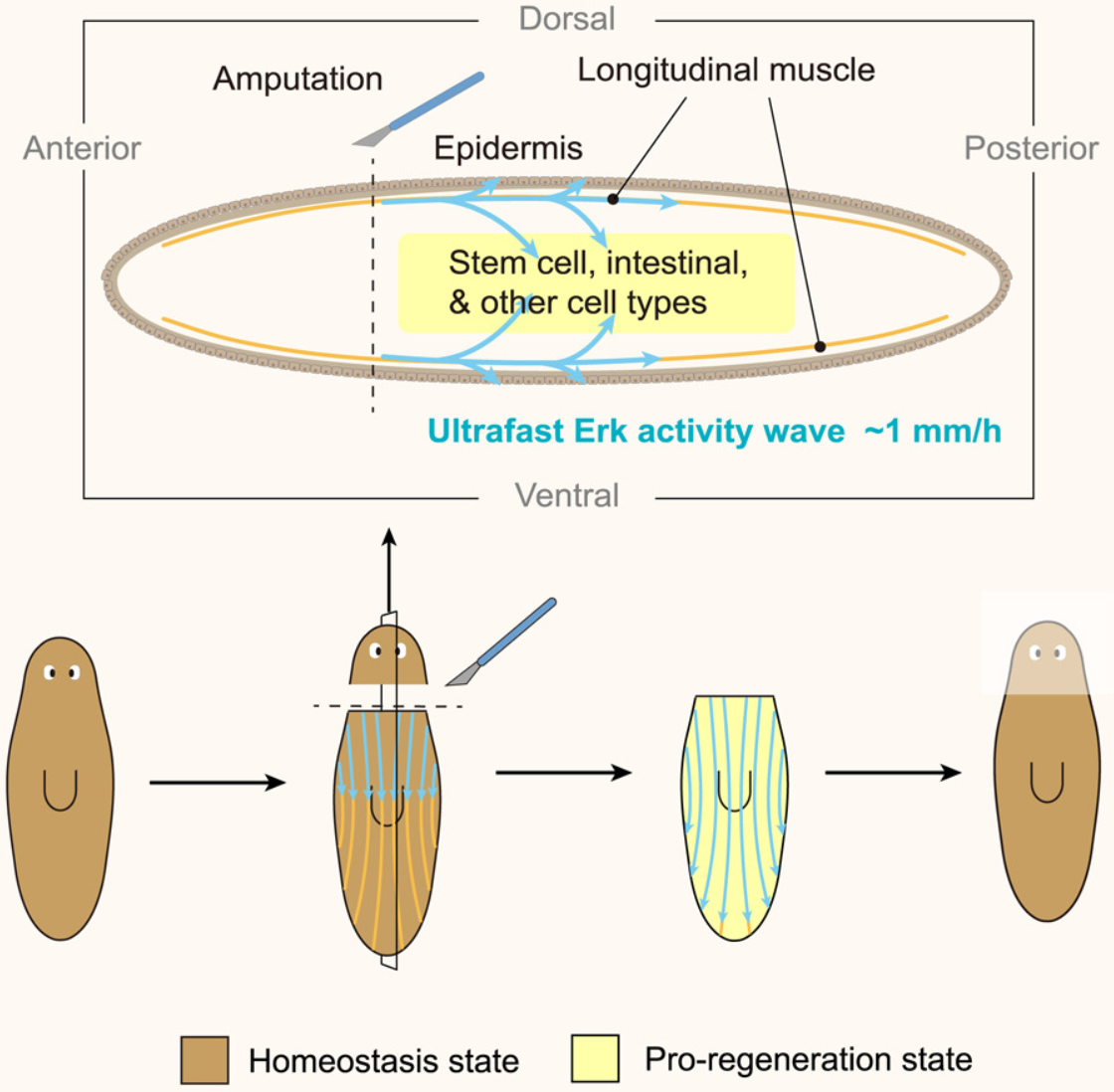
- 损伤会引起全身反应,但其功能仍然难以捉摸。快速同步远距离伤口反应的机制也大多未知。
- 利用具有全身再生能力的涡虫扁虫,我们发现损伤会诱导细胞外信号调节激酶 (Erk) 活性波的传播速度比其他多细胞组织快 10-100 倍。这种超快的传播需要纵向的体壁肌肉,细长的细胞形成密集的平行轨道,贯穿生物体的长度。肌肉的形态特性使它们能够充当传播和传播伤口信号的高速公路。
- 抑制 Erk 传播可以防止远离伤口的组织做出反应并阻止再生,这可以通过在第一次损伤后不久对远端组织进行第二次损伤来挽救。
- 我们的研究结果提供了一种在大型复杂组织中进行长距离信号传播的机制,以协调跨细胞类型的反应,并强调全身再生过程中空间分离的组织之间的反馈功能。
六体-INO80复合物揭示了非典型核小体重塑的结构基础
Hexasome-INO80 complex reveals structural basis of noncanonical nucleosome remodeling. Science
系列研究
- H2A-H2B 组蛋白二聚体丢失是活跃转录基因的标志,但细胞机制如何在非规范核小体颗粒背景下发挥作用仍然很大程度上难以捉摸。
- 在这项工作中,我们报告了 INO80 复合物对腺苷 5′-三磷酸依赖性染色质重塑六体的结构机制。我们展示了 INO80 如何识别因 H2A-H2B 丢失而出现的六体的非规范 DNA 和组蛋白特征。大规模的结构重排将 INO80 的催化核心转变为独特的自旋重塑模式,而其核肌动蛋白模块仍与长段未包裹的接头 DNA 相连。直接感应暴露的 H3-H4 组蛋白界面会激活 INO80,独立于 H2A-H2B 酸性斑块。
- 我们的研究结果揭示了 H2A-H2B 的缺失如何使重塑者能够进入不同的、但尚未探索的能量驱动染色质调控层。
INO80 在六体上的重新定位揭示了机械多功能性的基础
Reorientation of INO80 on hexasomes reveals basis for mechanistic versatility. Science
系列研究
- 与其他染色质重塑剂不同,INO80 优先动员六体,六体可在转录过程中形成。为什么 INO80 更喜欢六体而不是核小体仍不清楚。
- 在这里,我们报告了与六体或核小体结合的酿酒酵母 INO80 的结构。 INO80 以截然不同的方向结合两种底物。在六体上,INO80 将其 ATP 酶亚基 Ino80 置于超螺旋位置 –2 (SHL –2),这与之前在核小体上看到的 SHL –6 和 SHL –7 不同。
- 我们的结果表明,INO80 对六体的作用类似于其他重塑剂对核小体的作用,因此 Ino80 在 SHL –2 附近活性最大。 SHL –2 位置对于 INO80 的核小体重塑也起着关键作用。
- 总体而言,INO80 用于优先六体滑动的机械适应意味着亚核小体颗粒发挥着相当大的调节作用。
记录脑部微米级脉管系统的超柔性血管内探针
Ultraflexible endovascular probes for brain recording through micrometer-scale vasculature. Science
听上去很牛呀
美国斯坦福大学-化学工程系和生物工程系
- 植入式神经电子接口使神经系统疾病的基础研究和治疗取得了进展,但传统的颅内深度电极需要进行侵入性手术才能放置,并且在植入过程中可能会破坏神经网络。
- 我们开发了一种超小型、灵活的血管内神经探针,可以植入啮齿类动物大脑中 100 微米以下的血管中,而不会损坏大脑或脉管系统。在皮层和嗅球中选择性地实现了局部场电位和单个尖峰的体内电生理学记录。组织界面的组织学分析显示出最小的免疫反应和长期稳定性。
- 该平台技术可以轻松扩展为用于检测和干预神经系统疾病的研究工具和医疗设备。
SARS-CoV-2 Omicron BA.1 侵袭动态的基因组评估
Genomic assessment of invasion dynamics of SARS-CoV-2 Omicron BA.1. Science
英国牛津大学生物学系
- 严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 相关变种 (VOC) 现在是在人类连通性和群体免疫异质性的背景下出现的。
- 通过对 115,622 个 Omicron BA.1 基因组进行大规模系统动力学分析,我们确定了超过 6,000 次抗原不同的 VOC 进入英格兰的情况,并分析了它们的本地传播和扩散历史。我们发现,当 Omicron 在南部非洲首次报道时(2021 年 11 月 22 日),英国最大的 8 个 Omicron 谱系中的 6 个已经在传播。多个数据集显示,尽管随后由于从交通便利的次要地点出口而限制了来自南部非洲的旅行,但 Omicron 的进口仍在继续。 Omicron 传播谱系在英国的发起和传播是一个两个阶段的过程,可以通过该国的人文地理和分层旅行网络模型来解释。
- 我们的结果可以对推动 Omicron 和其他 VOC 在多个空间尺度上入侵的过程进行比较。
免疫介导的松果体去神经支配是心脏病睡眠障碍的基础
Immune-mediated denervation of the pineal gland underlies sleep disturbance in cardiac disease. Science
- 生理睡眠-觉醒周期的破坏和褪黑激素水平低经常伴随心脏病,但潜在的机制仍然是个谜。
- 对患有心脏病的人类和小鼠的光学透明松果体中的交感神经轴突进行免疫染色,结果显示与对照组相比,它们的去神经支配明显。
- 空间、单细胞、核和大量 RNA 测序将这种缺陷追溯到颈上神经节 (SCG),它通过炎症巨噬细胞的积累、纤维化和松果体支配神经元的选择性丧失来应对心脏病。SCG 中巨噬细胞的消耗可以防止与疾病相关的松果体去神经支配,并恢复褪黑激素的生理分泌。
- 我们的数据确定了心脏病中昼夜节律受到干扰的机制,并提出了治疗干预的目标。
生活环境微生物促进果蝇抗菌肽的特异性进化
Ecology-relevant bacteria drive the evolution of host antimicrobial peptides in Drosophila. Science
很有趣的研究。生活微生物与宿主基因演化
- 抗菌肽 (AMP) 是宿主编码的免疫效应物,其最初特征是其在对抗感染中的作用。 AMP 对于确定植物和动物宿主微生物组的组成也很重要。尽管许多研究表明 AMP 的快速进化,但对于驱动这种进化的选择压力却知之甚少。
- 宿主微生物组应对宿主免疫分子施加巨大的选择压力,因为宿主必须与其微生物伙伴保持微妙的平衡。正如最近对不同类群的调查所表明的那样,单个 AMP 的变化可能会破坏这种平衡。在果蝇中,先前的研究表明 AMP 家族双翅菌素 (Dpt) 进化迅速,包括 DptA 的氨基酸多态性 S69R 对宿主防御机会性病原体普罗威登斯菌 (Providencia rettgeri) 和普罗威登斯菌 (Providencia spp) 的主要影响。在缺乏多个 AMP 基因家族的果蝇中,宿主微生物组的有益细菌也会生长失控,尤其是肠道共生醋杆菌。果蝇物种编码两个双翅菌素基因,DptA 和 DptB,它们是源自 DptB 样基因的祖先复制的产物。为了检验宿主免疫库可能是专门进化来控制常见微生物组细菌的想法,我们筛选了最近制造的果蝇 AMP 突变体,用于防御醋杆菌属的感染。以确定是否有任何 AMP 基因可以解释果蝇如何控制这种互利共生的微生物。
- 我们发现单个 AMP 基因 DptB 解释了宿主抵抗多种醋杆菌属感染的能力。这种相互作用是高度特异性的:我们证实 DptA 不会有助于防御醋杆菌,而 DptB 不会有助于防御雷氏假单胞菌。因此,我们确定了双翅目双翅目的进化历史,并对果蝇和其他双翅目微生物组文献进行了系统回顾。我们意识到,在以水果为食的果蝇中,至少存在两次向 DptB 样基因趋同进化的事件,这是一种与高水平醋杆菌相关的生态学。这些观察结果表明,DptB 的进化是为了控制以水果为食的果蝇祖先中的醋杆菌。此外,继而采用蘑菇喂养生态的苍蝇一再丢失其 DptB 基因,同时蘑菇繁殖地也缺乏醋杆菌。类似的进化模式也出现在已经形成植物寄生生态的果蝇中,这些生态已经失去了 DptA 和 DptB 基因,并且具有缺乏普罗维登斯菌和醋杆菌的生态。为了研究这些 AMP 微生物特异性是否在整个果蝇中共享,我们用 DptA 和 DptB 样基因和等位基因的不同补充物感染了整个系统发育的物种。我们纳入了具有多种 DptA 样基因的物种,以及带有或不带有 DptB 的黑腹果蝇和食用蘑菇的果蝇。仅使用 DptA 或 DptB 的存在和多态性状态就可以轻松预测宿主对雷氏假单胞菌和醋酸杆菌感染的抵抗力,即使是在进化时间相隔约 5000 万年的果蝇物种中也是如此。
- 我们的研究展示了两种微生物特异性防御是如何由于产生两个双翅菌素基因的祖先复制而进化的。我们描述了一种单方面的进化动态,其中宿主使其免疫系统适应环境微生物,而不是宿主和微生物的共同进化。这一发现有助于解释跨类群 AMP 基因家族常见的快速进化爆发背后的进化逻辑。我们的结果还揭示了为什么某些 AMP 在防御特定微生物方面可以发挥如此不成比例的作用:它们是为此目的而进化选择的。这一认识表明,基因组可以编码“残余”免疫效应器,AMPs是为了防御不再与宿主现代生态相关的微生物而进化的。因此,微生物特异性效应子的产生和丧失为免疫系统提供了一种高效的机制,用于定制宿主防御以控制生态相关微生物。

人类骨骼形态的遗传结构和进化
The genetic architecture and evolution of the human skeletal form. Science
一个我特别想了解的研究问题
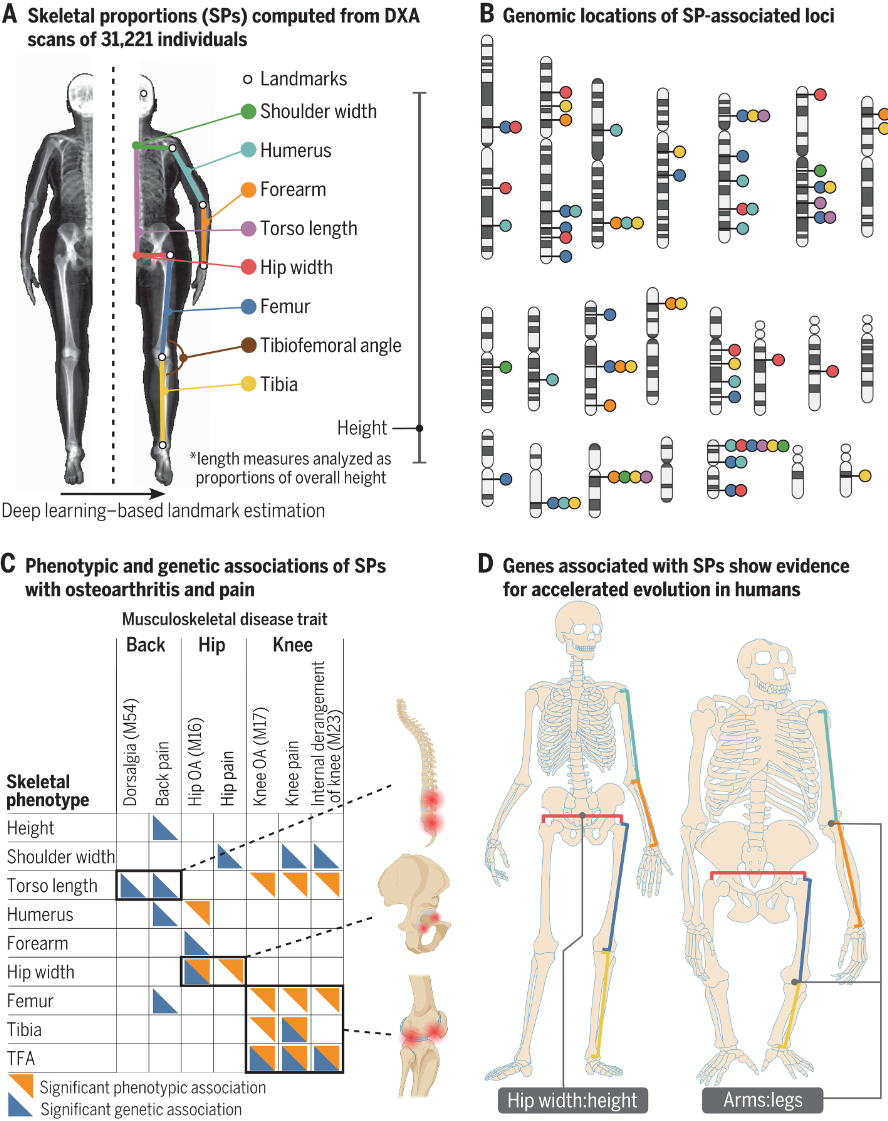
- 由于我们独特的骨骼形态,人类是唯一的双足类人猿。古人类学对影响骨骼形态的形态变化进行了广泛的研究。由于样本量有限,除了站立高度之外,检查个体骨骼差异和特定生长及其进化的遗传基础一直具有挑战性。
- 研究骨骼形态的一种方法是获取基因组中影响骨骼发育和形态的区域图谱。此前,主要通过动物模型和比较基因组学对此进行了研究,但这些方法的通量基本上较低。一种补充方法是检查人类骨骼特征变异的遗传基础。
- 在这项工作中,我们将深度学习模型应用于来自英国生物银行的 31,221 幅全身双能 X 射线吸收测定 (DXA) 图像,以提取 23 种不同的图像衍生表型,其中包括所有长骨长度以及臀部和肩部宽度,我们在控制高度的同时对这些表型进行分析。
- 所有骨骼比例 (SP) 均具有高度遗传性(约 30% 至 50%),这些性状的全基因组关联研究确定了 145 个独立基因座。这些基因座富含调节骨骼发育的基因以及与罕见的人类骨骼疾病和异常小鼠骨骼表型相关的基因。遗传相关性和基因组结构方程模型表明,肢体比例表现出很强的遗传共享性,但在遗传上与宽度和躯干比例无关。表型和多基因风险评分分析确定了髋关节和膝关节骨关节炎(美国成人残疾的主要原因)与相应地区的 SP 之间的特定关联。我们还发现了人类手臂到腿和臀部宽度比例进化变化的基因组证据,与古人类化石记录中这些 SP 的显着解剖学变化一致。与心血管、自身免疫、代谢和其他类别的性状相比,与这些 SP 相关的基因座在人类加速区域和在人类和类人猿整个发育过程中差异表达的基因调控元件中显着富集。
- 我们的工作验证了在 DXA 图像上使用深度学习模型来识别影响人类骨骼形态的特定遗传变异。它还将人类解剖学变化的一个主要进化方面与发病机制联系起来。
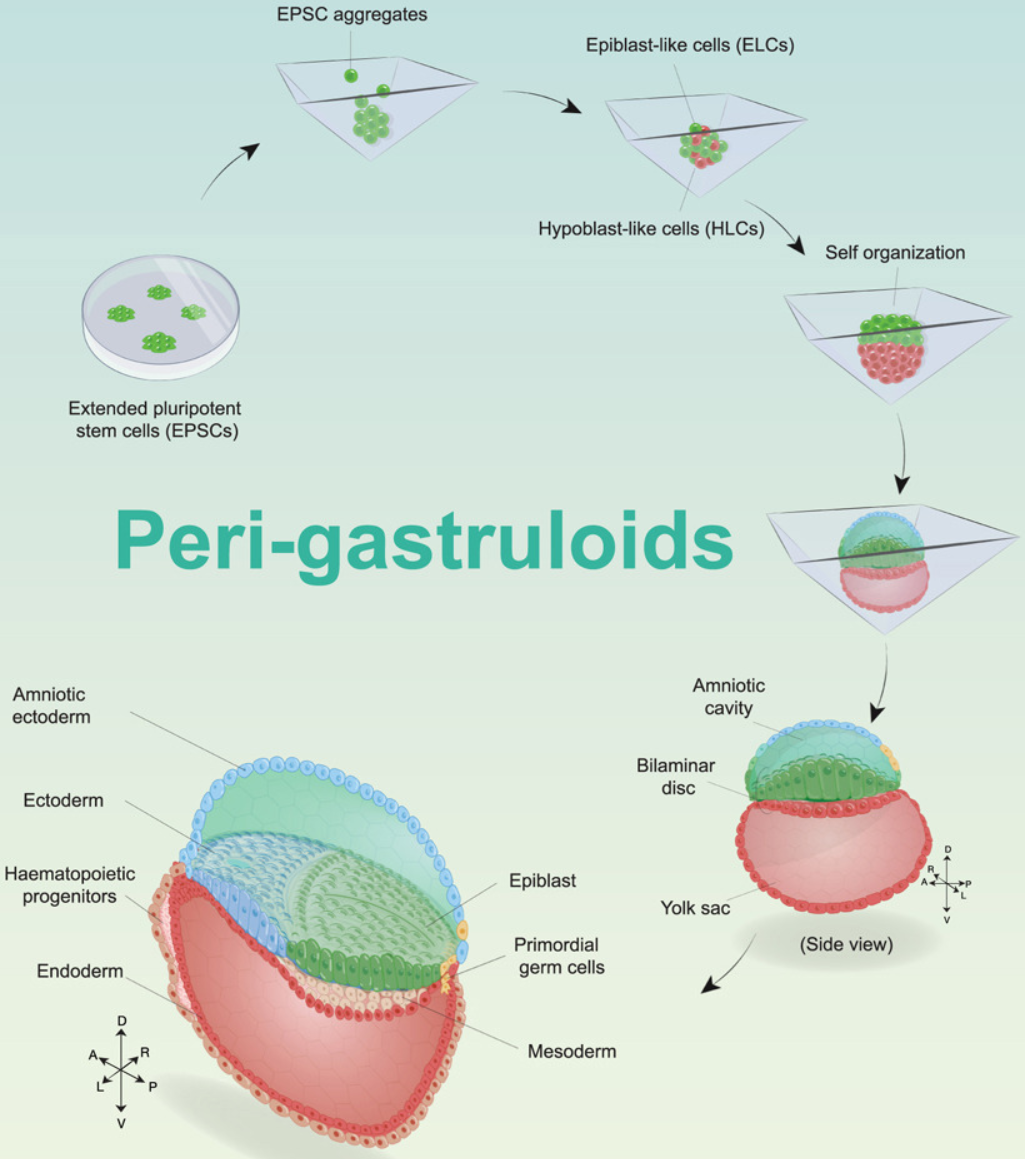
人类干细胞衍生的类原肠胚结构
德克萨斯大学西南医学中心分子生物学系
所有技术有什么特别之处?
- 复制人类原肠胚形成的体外干细胞模型已经建立,但它们缺乏胚胎发育、形态发生和模式形成所需的必需胚胎外细胞。
- 在这里,我们描述了一种强大而有效的方法,可促使人类扩展多能干细胞自组织成胚胎样结构,称为环类原肠胚,包括胚胎(外胚层)和胚外(下胚层)组织。尽管由于滋养细胞的排除,原肠胚周围细胞无法存活,但它们概括了人类原肠胚形成周围发育的关键阶段,例如形成羊膜和卵黄囊腔、发育二层和三层胚胎盘、指定原始生殖细胞、启动原肠胚形成以及经历早期神经形成和器官发生。
- 单细胞 RNA 测序揭示了高级人类原肠胚周围细胞类型与人类和非人类灵长类动物中发现的初级原肠胚周围细胞类型之间的转录组相似性。
- 这种原肠胚周围平台允许对原肠胚形成之外的进一步探索,并可能有助于开发用于再生医学的人类胎儿组织。
肿瘤耐药单细胞克隆的异质性
Diverse clonal fates emerge upon drug treatment of homogeneous cancer cells. Nature
芝加哥-西北大学-范伯格医学院-细胞与发育生物学系
部分地回答了我一直想了解的问题。理论上,基于单细胞的耐药机制探索应该可以非常高效地研究药物靶点和耐药机制。
- 即使在基因相同的癌细胞中,对治疗的抵抗也经常出现在这些细胞的一小部分中。初始群体中稀有单个细胞的分子差异使某些细胞对治疗产生耐药性;然而,人们对耐药结果的变异性知之甚少。
- 在这里,我们开发并应用 FateMap,这是一个将 DNA 条形码与单细胞 RNA 测序相结合的框架,以揭示数十万接受抗癌治疗的克隆的命运。我们发现,单细胞来源的癌细胞中出现的耐药克隆在分子、形态和功能上都具有不同的耐药类型。这些耐药类型很大程度上是由添加药物之前细胞之间的分子差异决定的,而不是由外在因素决定的。
- 药物剂量和类型的变化可以改变初始细胞的耐药类型,导致某些耐药类型的产生和消除。来自患者的样本显示了临床背景下这些耐药类型存在的证据。
- 我们观察到几种单细胞来源的癌细胞系和用多种药物治疗的细胞类型的耐药类型的多样性。由于内在细胞状态的可变性而导致的耐药类型的多样性可能是对外部线索反应的一般特征。
cDNA 展示蛋白水解
Mega-scale experimental analysis of protein folding stability in biology and design. Nature
挺特别的研究
- DNA 测序和机器学习的进步正在为大规模蛋白质序列和结构提供见解。然而,驱动折叠的能量在这些结构中是不可见的,并且在很大程度上仍然未知。折叠的隐藏热力学可以驱动疾病,塑造蛋白质进化并指导蛋白质工程,并且需要新的方法来揭示每个序列和结构的这些热力学。
- 在这里,我们介绍了 cDNA 展示蛋白水解,这是一种在一周的实验中测量多达 900,000 个蛋白质结构域的热力学折叠稳定性的方法。从总共 180 万次测量中,我们策划了一组约 776,000 个高质量折叠稳定性,涵盖所有单氨基酸变体以及 331 个天然和 148 个长度为 40-72 个氨基酸的从头设计蛋白质结构域的选定双突变体。
- 使用这个广泛的数据集,我们量化了(1)影响氨基酸适应性的环境因素,(2)蛋白质位点之间的热力学耦合(包括意外的相互作用),以及(3)进化氨基酸使用和蛋白质折叠稳定性之间的整体差异。我们还研究了我们的方法如何识别设计蛋白质中的稳定性决定因素并评估设计方法。
- cDNA展示蛋白水解方法快速、准确且具有独特的可扩展性,有望揭示氨基酸序列如何编码折叠稳定性的定量规则。
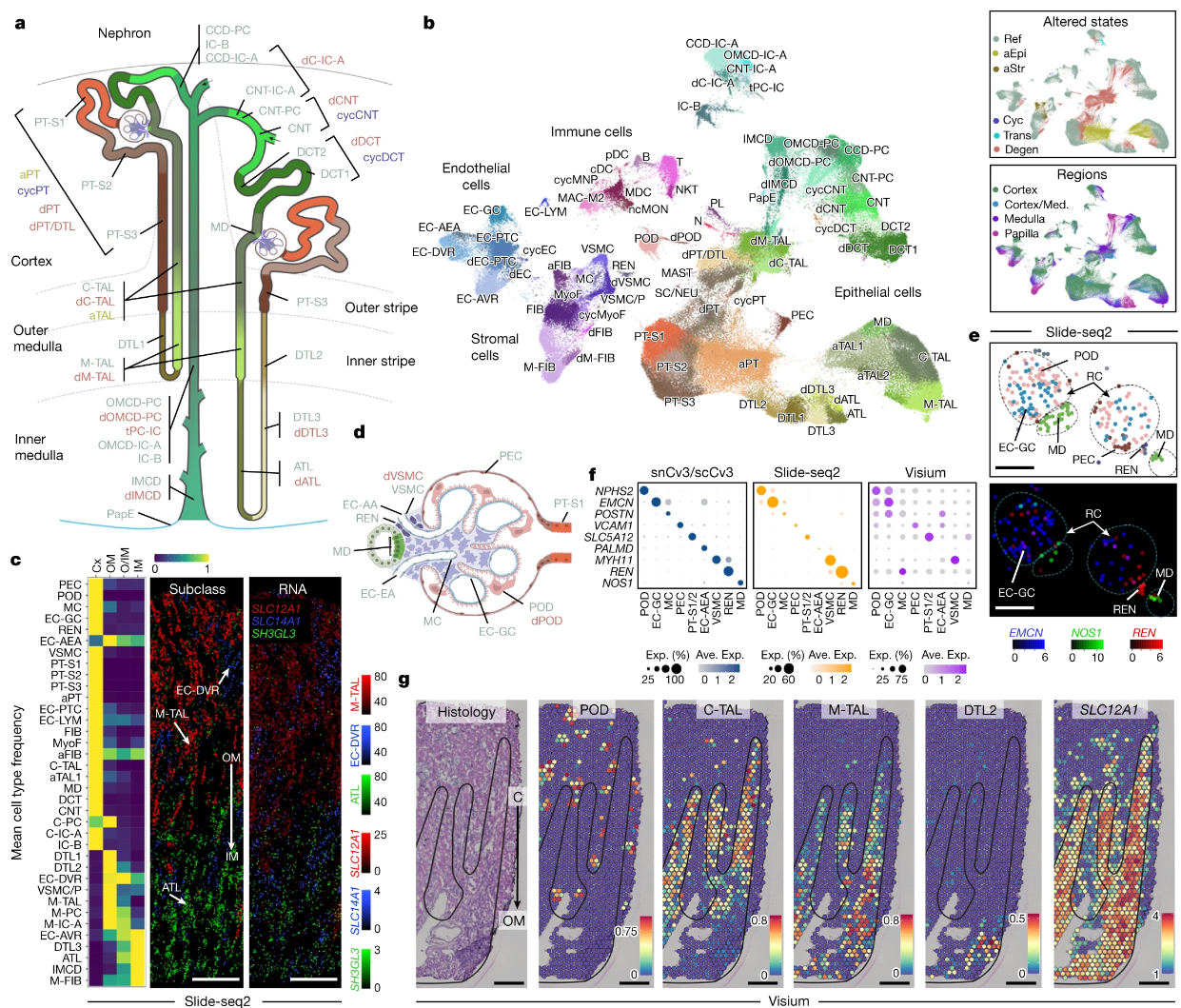
人类肾脏中健康和受损细胞状态和生态位的图谱
An atlas of healthy and injured cell states and niches in the human kidney. Nature. full html
- 了解肾脏疾病依赖于定义细胞类型和状态的复杂性、其相关的分子特征以及组织邻域内的相互作用。
- 在这里,我们将多种单细胞和单核测定(> 400,000 个细胞核或细胞)和空间成像技术应用于广泛的健康参考肾脏(45 个供体)和患病肾脏(48 个患者)。这提供了 51 种主要细胞类型的高分辨率细胞图谱,其中包括罕见的和以前未描述的细胞群。多组学方法提供了整个肾脏的详细转录组学概况、调控因素和空间定位。
- 我们还定义了肾单位段和间质中在肾损伤时发生改变的 28 种细胞状态,包括循环状态、适应性(成功或适应不良修复)、过渡和退行性状态。分子特征允许使用空间转录组学在损伤邻域内定位这些状态,而大规模 3D 成像分析(约 120 万个邻域)提供了与主动免疫反应的相应联系。这些分析定义了与损伤时程和生态位相关的生物途径,包括预测与肾功能下降相关的适应不良状态的上皮修复的特征。
- 这种健康和患病人类肾脏的综合多模态空间细胞图谱代表了细胞状态、邻域、结果相关特征和公开的交互式可视化的综合基准。
DNA复制中Pol II相关的转录激活
RNA polymerase II associates with active genes during DNA replication. Nature
- 转录机制被认为在复制过程中与 DNA 分离。某些蛋白质(称为表观遗传标记)必须从亲本 DNA 链转移到子代 DNA 链,以维持转录状态的记忆。这些蛋白质被认为会重新启动染色质结构的重建,最终将 RNA 聚合酶 II (Pol II) 募集到新复制的子链上。据信,只有在染色质重建后,Pol II 才会被招募回活性基因。然而,几乎没有实验证据来解决 Pol II 何时以及如何被招募回子链并恢复转录的核心问题。
- 在这里,我们表明,复制叉通过后,Pol II 与其他一般转录蛋白和未成熟 RNA 复合,立即与新生 DNA 前导链和滞后链上的活性基因重新关联,并快速恢复转录。这表明具有转录活性的 Pol II 复合物保留在 DNA 附近,而 Pol II-PCNA 相互作用可能是这种保留的基础。
- 这些发现表明,在从 DNA 复制到恢复转录的过渡过程中,Pol II 机制可能不需要将表观遗传标记招募到新合成的 DNA 中。
HLA-B基因座与SARS-CoV-2的预T细胞免疫
A common allele of HLA is associated with asymptomatic SARS-CoV-2 infection. Nature
- 研究表明,至少 20% 的 SARS-CoV-2 感染者仍然无症状。尽管全球大多数努力都集中在 COVID-19 的严重疾病上,但检查无症状感染提供了一个独特的机会来考虑促进病毒快速清除的早期免疫学特征。
- 在这里,假设人类白细胞抗原 (HLA) 基因座的变异可能是介导无症状感染的潜在过程,我们在一项旨在追踪 COVID-19 症状的基于智能手机的研究中招募了 29,947 名个体,这些人可以获得高分辨率的 HLA 基因分型数据和结果。我们的发现队列(n = 1,428)由报告 SARS-CoV-2 检测结果呈阳性的未接种疫苗的个体组成。
- 我们测试了五个 HLA 基因座与病程的关联,并在两个独立队列中观察到,HLA-B*15:01 与无症状感染之间存在很强的关联。我们表明,这种遗传关联是由于预先存在的 T 细胞免疫所致,我们发现,来自携带 HLA-B*15:01 的个体的大流行前样本中的 T 细胞对免疫显性 SARS-CoV-2 S 衍生肽 NQKLIANQF 有反应。大多数反应性 T 细胞表现出记忆表型,具有高度多功能性,并且与季节性冠状病毒衍生的肽发生交叉反应。 HLA-B*15:01-肽复合物的晶体结构表明,肽 NQKLIANQF 和 NQKLIANAF(来自 OC43-CoV 和 HKU1-CoV)具有相似的被 HLA-B*15:01 稳定和呈递的能力。最后,我们证明肽的结构相似性支撑了高亲和力公共 T 细胞受体的 T 细胞交叉反应性,为 HLA-B*15:01 介导的预先存在的免疫提供了分子基础。
生物分子凝聚体的极端动力学
- 蛋白质和核酸可以在细胞中发生相分离,形成浓缩的生物分子凝聚物。冷凝物的功能跨越许多长度尺度:它们在分子尺度上调节相互作用和化学反应,在介观尺度上组织生化过程,并划分细胞。了解这些过程的根本机制需要详细了解这些尺度的丰富动态。生物分子凝聚体的介观动力学已被广泛表征,但它们在分子尺度上的行为仍然更加难以捉摸。
- 在这里,作为生物分子相分离的一个例子,我们研究了两种高度带相反电荷的无序人类蛋白质的复杂凝聚层。它们的密相比稀相浓度高 1,000 倍,由此产生的渗透相互作用网络导致体积粘度比水大 300 倍。
- 然而,针对单个液滴内的测量而优化的单分子光谱表明,在分子尺度上,无序蛋白质仍然非常动态,它们的链构型在亚微秒时间尺度上相互转换。大规模全原子分子动力学模拟再现了实验观察结果并解释了这种明显的差异:各个带电侧链之间的潜在相互作用是短暂的,并且在皮秒到纳秒的时间尺度上进行交换。
- 我们的结果表明,尽管相分离系统的宏观粘度很高,但分子尺度上有效反应所需的局部生物分子重排仍然可以快速进行。
肺泡 I 型上皮细胞与肺癌发生
KRAS(G12D) drives lepidic adenocarcinoma through stem-cell reprogramming. Nature
系列研究
- 许多癌症起源于被驱动复制的体细胞突变劫持的干细胞或祖细胞,例如肺泡上皮 II 型 (AT2) 细胞的腺瘤转化。
- 在这里,我们演示了一种不同的情况:分化的 AT1 细胞中 KRAS(G12D) 的表达将它们缓慢且异步地重新编程回 AT2 干细胞,从而继续生成惰性肿瘤。与人类鳞状腺癌一样,肿瘤细胞以非破坏性方式沿着肺泡壁缓慢扩散,并且 ERK 活性较低。我们发现 AT1 和 AT2 细胞作为不同的起源细胞,对同时的 WNT 激活和 KRAS(G12D) 诱导表现出不同的反应,这加速了 AT2 衍生的腺瘤增殖,但抑制了 AT1 衍生的腺瘤增殖。 KRAS(G12D) 诱导的 AT1 细胞中 ERK 活性的增强可提高转化效率、增殖以及从鳞状肿瘤组织学向混合肿瘤组织学的进展。
- 总的来说,我们已经确定了一种新的肺腺癌起源细胞,即 AT1 细胞,它概括了人类鳞状癌的特征。在此过程中,我们还发现了致癌 KRAS 在腺瘤转化过程中将分化的静止细胞重新编程回其亲本干细胞的能力。我们的工作进一步揭示,无论特定癌症当前的分子特征和驱动癌基因如何,起源细胞都会对其后续行为产生普遍而持久的影响。
肺泡 I 型上皮细胞与肺癌发生
p53 governs an AT1 differentiation programme in lung cancer suppression . Nature
系列研究
- 肺癌是全球癌症死亡的主要原因。肿瘤抑制基因 TP53 的突变发生在 50% 的肺腺癌 (LUAD) 中,并与不良预后相关,但 p53 如何抑制 LUAD 的发展仍然是个谜。
- 我们在此表明 p53 通过控制细胞状态,特别是通过促进肺泡 1 型 (AT1) 分化来抑制 LUAD。使用在肺泡 2 型 (AT2) 细胞中表达致癌 Kras 和无效、野生型或超态 Trp53 等位基因的小鼠,我们观察到 p53 对 LUAD 启动和进展的分级影响。 LUAD 细胞的 RNA 测序和 ATAC 测序发现,在体内肿瘤抑制过程中,p53 通过直接 DNA 结合、染色质重塑和诱导 AT1 细胞特征基因来诱导 AT1 分化程序。单细胞转录组学分析表明,在 LUAD 进化过程中,p53 通过过渡细胞状态的作用促进 AT1 分化,类似于肺泡损伤修复中 AT2 到 AT1 细胞分化过程中看到的短暂中介。
- 值得注意的是,p53 失活会导致这些过渡性癌细胞的不适当持续存在,并伴有生长信号上调以及与肺谱系身份(与 LUAD 进展相关的特征)的分歧。对 Trp53 野生型和 Trp53 缺失小鼠的分析表明,p53 还通过调节 AT2 细胞自我更新和促进过渡细胞分化为 AT1 细胞来指导损伤后的肺泡再生。
- 总的来说,这些发现阐明了 p53 介导的 LUAD 抑制机制,其中 p53 控制肺泡分化,并表明肿瘤抑制反映了 p53 在协调损伤后组织修复中的基本作用。
人类母胎界面的空间解析时间轴
A spatially resolved timeline of the human maternal–fetal interface. Nature
- 从妊娠早期开始,胎儿来源的绒毛外滋养细胞 (EVT) 侵入子宫并重塑其螺旋动脉,将其转变为大而扩张的血管。已经提出了几种机制来解释 EVT 如何与母体蜕膜协调以促进有利于螺旋动脉重塑 (SAR) 的组织微环境。然而,关于哪些免疫细胞和基质细胞参与这些相互作用以及这种相互作用如何随胎龄变化仍然存在争议。
- 在这里,我们使用多组学方法,结合空间蛋白质组学和转录组学的优势,构建了妊娠前半期人类母胎界面的时空图谱。我们使用飞行时间多重离子束成像和 37 重抗体组来分析 66 名妊娠 6 至 20 周的个体完整蜕膜内的约 500,000 个细胞和 588 个动脉,并将该数据集与共同注册的转录组学概况相结合。孕龄显着影响母体免疫和基质细胞的频率,表达 CD206、CD163、TIM-3、galectin-9 和 IDO-1 的耐受亚群在以后的时间点变得越来越丰富和共定位。相比之下,SAR 进展优先与 EVT 侵袭相关,并由 78 个基因本体通路转录定义,表现出明显的单相和双相趋势。
- 最后,我们开发了一个 SAR 综合模型,其中侵袭伴随着促血管生成、免疫调节 EVT 程序的上调,这些程序促进与血管内皮的相互作用,同时避免母体免疫细胞的激活。
OBOX 调节小鼠合子基因组激活和早期发育
OBOX regulates murine zygotic genome activation and early development. Nature
- 合子基因组激活 (zygotic genome activation, ZGA) 激活静止基因组以实现母体到合子的转变。然而,哺乳动物体内 ZGA 背后的转录因子 (TF) 的身份仍然难以捉摸。
- 在这里,我们证明了 OBOX,一个类似 PRD 的同源盒结构域 TF 家族 (OBOX1-8),是小鼠 ZGA 的关键调节因子。母源转录的 Obox1/2/5/7 和合子表达的 Obox3/4 缺陷的小鼠出现 2-4 细胞停滞,并伴有 ZGA 受损。母体和合子 OBOX 冗余地支持胚胎发育,因为 Obox KO 缺陷可以通过恢复其中任何一个来挽救。染色质结合分析显示 Obox 敲除优先影响 OBOX 结合靶标。
- 从机制上讲,OBOX 促进了 RNA Pol II 的“预配置”,因为 Pol II 从最初的 1 细胞结合靶标重新定位到 ZGA 基因启动子和远端增强子。 Obox 突变体中受损的 Pol II 预配置伴随着有缺陷的 ZGA 和染色质可及性转变,以及 1 细胞 Pol II 靶标的异常激活。最后,OBOX 的异位表达激活了 mESC 中的 ZGA 基因和 MERVL 重复序列。
- 因此,这些数据表明 OBOX 调节小鼠 ZGA 和早期胚胎发生。
肿瘤免疫类
抗原和背景信息的共同转移协调外周和淋巴结常规树突状细胞激活
免疫协调的远程调控
- 针对感染和癌症的 T 细胞反应是由远离攻击部位的淋巴结中的传统树突状细胞 (cDC) 指导的。从组织迁移到淋巴结的迁移性 cDC 不仅驱动初始 T 细胞激活,还将抗原转移到淋巴结驻留的 cDC。这些驻留细胞在定义所产生的 T 细胞反应的特征方面发挥着重要作用;然而,鉴于它们的空间分离,尚不清楚它们如何适当地处理和呈递抗原以适当地直接反应。
- 在这里,我们使用新型甲型流感毒株和改良的黑色素瘤模型,表明组织和淋巴结 cDC 激活是协调的,并且这是由上下文线索的共同转移驱动的。在肿瘤中,肿瘤微环境中不完全的 cDC 激活由淋巴结驻留的 cDC 反映出来,而在流感感染期间,病原体相关分子模式与抗原共同转移,驱动驻留 cDC 中的 TLR 信号传导及其随后的强烈激活。这种共转移机制解释了驻留 cDC 如何独特地处理单个抗原,以及驱动肿瘤 cDC 激活不良的信号如何进一步影响淋巴结。
- 我们的研究结果阐明了组织环境如何决定抗原,从而决定淋巴结中 T 细胞的命运。
临床类
基于 Epstein-Barr 病毒的鼻咽癌筛查的性能和操作可行性:两种替代方法的直接比较
- 两种基于 Epstein-Barr 病毒 (EBV) 的检测方法已显示出早期检测鼻咽癌 (NPC) 的前景。两者都没有经过独立验证,也没有对它们的性能进行比较。我们比较了他们在独立人群中的诊断表现。
- 我们检测了 2010 年至 2014 年诊断的 819 例台湾鼻咽癌病例(213 例早期,美国癌症联合委员会第 7 版 I 期和 II 期)和来自同一地区的 1,768 例对照者的血液样本,频率与年龄和性别的病例相匹配。我们比较了通过ELISA法测量的免疫球蛋白 A 抗体的 EBV 抗体评分(EBV 抗体评分)和通过实时 PCR 测量的血浆 EBV DNA 负荷,然后在 EBV DNA 阳性个体中进行下一代测序 (NGS)(EBV DNA 算法)。
- 分别测量了 2,522 名(802 例病例;1,720 名对照)和 2,542 名(797 名病例;1,745 名对照)个体的 EBV 抗体和 DNA 载量。在 898 名血浆 EBV DNA 呈阳性并因此符合 NGS 资格的个体中,我们选择了 442 名 (49%) 进行 NGS 检测。 EBV 抗体评分对 NPC 的敏感性为 88.4%(95% CI,86.1 至 90.6),特异性为 94.9%(95% CI,93.8 至 96.0)。 EBV DNA 算法具有显着更高的灵敏度(93.2%;95% CI,91.3 至 94.9;P = 1.33 × 10-4)和特异性(98.1%;95% CI,97.3 至 98.8;P = 3.53 × 10-7)。对于早期 NPC,EBV 抗体评分的敏感性为 87.1%(95% CI,82.7 至 92.4),EBV DNA 算法的敏感性为 87.0%(95% CI,81.9 至 91.5)(P = .514)。对于鼻咽癌发病率为20-100/100,000人年的地区(例如中国南方和香港的居民),这两种方法得到的筛查所需数字相似(EBV抗体评分:5,656-1,131;EBV DNA算法:5,365-1,073);阳性预测值分别为 0.4% 至 1.7% 和 1.0% 至 4.7%。
- 我们证明了 EBV 抗体和血浆 EBV DNA 用于鼻咽癌检测的高灵敏度和特异性,但 EBV 抗体评分的表现稍差。需要进行成本效益研究来指导筛查的实施。
食管癌或胃食管结合部癌新辅助化疗或放化疗的个体参与者数据网络荟萃分析
- 目的:胸段食管(TE)或胃食管交界处(GEJ)可切除癌的最佳新辅助治疗仍然存在争议。我们对随机对照试验 (RCT) 进行了个体参与者数据 (IPD) 网络荟萃分析 (NMA),以研究化疗或放化疗的效果,重点关注肿瘤位置和组织学亚组。
- 患者和方法:所有已发表或未发表的随机对照试验在 2015 年 12 月 31 日之前结束累积,并且比较了以下至少两种策略均符合资格:前期手术 (S)、化疗后手术 (CS) 以及放化疗后进行手术(CRS)。所有分析都是根据研究人员获得的 IPD 进行的。主要终点是总生存期(OS)。 IPD-NMA 通过针对年龄、性别、肿瘤位置和组织学进行调整的一步混合效应 Cox 模型进行分析。 NMA 在 PROSPERO 中注册 (CRD42018107158)。
- 结果:35 项 RCT 中的 26 项(5,807 名患者中的 4,985 名)获得了 IPD,对应于 CS-S 的 12 项比较、CRS-S 的 12 项比较和 CRS-CS 的 4 项比较。与 S 相比,CS 和 CRS 导致 OS 增加,风险比 (HR) = 0.86(0.75 至 0.99),P = 0.03,HR = 0.77(0.68 至 0.87),P < 0.001。 NMA 比较 CRS 与 CS 的 OS,HR 为 0.90(0.74 至 1.09),P = .27(一致性 P = .26,异质性 P = .0038)。对于 CS 与 S,观察到 GEJ 肿瘤与 TE 肿瘤对 OS 的影响更大 (P = .036)。对于 CRS 与 S 以及 CRS 与 CS 相比,观察到女性对 OS 的影响更大(分别为 P = 0.003、0.012)。
- 结论:从组织学角度来看,新辅助化疗和放化疗始终优于单独 S 治疗,但 CRS 的治疗效果大小和 CS 肿瘤位置的治疗效果因性别而异。尚未发现 CS 和 CRS 之间存在显着的 OS 差异。
高PD-L1表达与 II-IIIA 期NSCLC患者的atezolizumab获益
- 背景:IMpower010 (NCT02486718) 证明,在 DFS 中期分析中,PD-L1 阳性和所有 II-IIIA 期非小细胞肺癌里,与铂类化疗后的最佳支持治疗 (BSC) 相比,辅助 atezolizumab 显着改善无病生存 (DFS) (NSCLC) 人群。本文报告了首次总生存期 (OS) 中期分析的结果。
- 患者和方法:这项 3 期、开放标签、1:1 随机研究的设计、受试者和主要终点 DFS 结局已报告,该研究是在辅助铂类化疗后进行的 atezolizumab(1200 mg q3w;16 个周期)与 BSC 的比较(1-4 个周期)用于完全切除的 IB 期(≥4 cm)-IIIA NSCLC 成人。关键的次要终点包括 IB-IIIA 期意向治疗 (ITT) 人群的 OS 和随机治疗患者的安全性。第一个预先指定的 OS 中期分析是在 ITT 人群中 251 例死亡后进行的。探索性分析包括基线 PD-L1 表达水平的 OS(SP263 测定)。
- 结果:在 2022 年 4 月 18 日平均 45.3 个月的随访中,atezolizumab 组的 507 名患者中有 127 名患者 (25%) 死亡,BSC 组的 498 名患者中有 124 名患者 (24.9%) 死亡。 ITT 人群的中位 OS 无法估计;分层 HR 为 0.995 (95% CI 0.78-1.28)。 II-IIIA 期 (n=882) 的分层 OS HR (95% CI) 为 0.95 (0.74-1.24),II-IIIA 期 PD-L1 TC ≥1% 的分层 OS HR (95% CI) 为 0.71 (0.49-1.03) (n= 476),II-IIIA 期 PD-L1 TC ≥50% (n=229) 人群中为 0.43 (95% CI 0.24-0.78)。自上次分析以来,Atezolizumab 相关不良事件发生率保持不变(495 名患者中,分别有 53 名患者 [10.7%] 为 3/4 级,4 名患者 [0.8%] 为 5 级)。
- 结论:尽管 ITT 人群的 OS 仍不成熟,但这些数据表明在 PD-L1 亚组分析中存在有利于 atezolizumab 的积极趋势,这主要是由 PD-L1 TC ≥ 50% II-IIIA 期亚组驱动的。 13 个月的额外随访后没有观察到新的安全信号。总之,这些发现支持了辅助阿替利珠单抗在这种情况下的积极获益-风险特征。
综合基因组分析与可靶向治疗的可能性
First-Line Genomic Profiling in Previously Untreated Advanced Solid Tumors for Identification of Targeted Therapy Opportunities. JAMA Netw Open. full html
日本京都京都大学医院临床肿瘤科
了解一下方法学。GSClassifier背景。
- 重要性:通过新一代测序进行综合基因组分析 (CGP) 的精准肿瘤学旨在伴随诊断和基因组分析。标准护理(SOC)之前CGP的临床效用尚未解决,需要更多证据。
- 目的:探讨下一代 CGP(FoundationOne CDx [F1CDx])在先前未经治疗的转移性或复发性实体瘤患者中的临床效用。
- 设计、环境和参与者:这项多中心、前瞻性、观察性队列研究招募了2021年5月18日至2022年2月16日期间既往未经治疗的晚期实体瘤患者,随访至2022年8月16日。该研究于2022年8月16日进行。日本有6家医院。符合条件的患者年龄为 20 岁或以上,东部肿瘤合作组表现状态为 0 至 1,且患有先前未经治疗的胃肠道或胆道转移性或复发性癌症;胰腺、肺、乳房、子宫或卵巢;和恶性黑色素瘤。暴露:在 SOC 之前对晚期实体瘤进行全面的基因组分析测试。主要结果和措施:具有可操作或可药物基因组改变和基于分子的推荐治疗(MBRT)的患者比例。
- 结果:共有 183 名患者符合纳入标准,其中 180 名患者(92 名男性 [51.1%]),中位年龄 64 岁(范围,23-88 岁)随后接受了 CGP(肺 [n = 28]、结肠/小肠 [n = 27]、胰腺 [n = 27]、乳房 [n = 25]、胆道 [n = 20]、胃 [n = 19]、子宫 [n = 12]、食道 [n = 10] 、卵巢 [n = 6] 和皮肤黑色素瘤 [n = 6])。 172 名患者的数据可用于终点分析。在 172 名患者中发现了可操作的改变(100.0%;95% CI,97.9%-100.0%),在 109 名患者中发现了可药物改变(63.4%;95% CI,55.7%-70.6%)。分子肿瘤委员会确定 105 名患者接受 MBRT(61.0%;95% CI,53.3%-68.4%)。在肿瘤不可知环境中的 49 名患者(28.5%;95% CI,21.9%-35.9%)中发现了 CGP 测试伴随诊断列表中包含的基因组改变。中位随访时间为 7.9 个月(范围为 0.5-13.2 个月)后,34 名患者(19.8%;95% CI,14.1%-26.5%)接受了 MBRT。
- 结论和相关性:本研究的结果表明,晚期实体瘤患者在 SOC 之前进行 CGP 检测可能在临床上有益于指导后续抗癌治疗,包括分子匹配治疗。
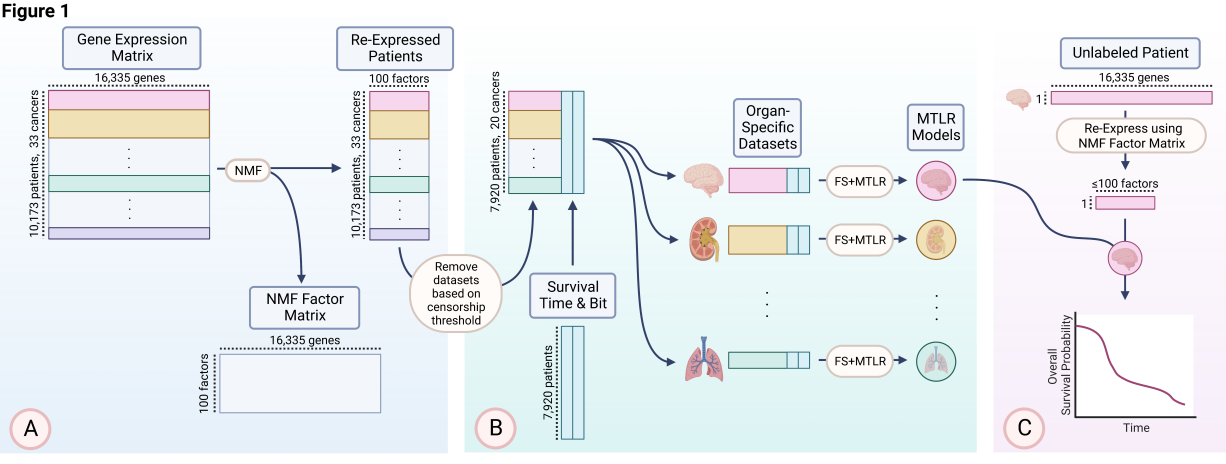
基于泛癌和 NMF-MTLR 计算肿瘤患者的生存概率
Learning Individual Survival Models from PanCancer Whole Transcriptome Data. Clin Cancer Res. full pdf
可作为GSClassifier背景
- 目的:个性化医疗试图根据每位患者的个体肿瘤分子特征来预测其生存时间。我们研究了我们的生存学习器与降维方法相结合是否可以为各种癌症患者产生有用的生存估计。
- 实验设计:本文提供了一种从 PanCancer Atlas 中学习预测个体癌症患者生存时间的模型的方法:给定 10173 名患者的(16335 维)基因表达谱,每名患者患有 33 种癌症中的一种,该方法使用无监督 NMF(非负矩阵分解)以 100 个学习的 NMF 因子重新表达每个患者的基因表达数据。然后,它将这 100 个因素输入 MTLR(多任务 Logistic 回归)学习器,为 20 种癌症中的每一种(具有超过 50 个未经审查的实例)生成癌症特定模型;这会产生“个体生存分布”(ISD),它提供每个患者未来每个时间的生存概率 – 提供患者的风险评分和估计生存时间。
- 结果:我们的 NMF-MTLR 一致性指数总体优于 VAECox 基准 14.9%。我们使用泛癌 NMF 结合癌症特异性 MTLR 模型实现了最佳生存预测。我们提供 NMF 模型的生物学解释以及 ISD 对预后和治疗反应预测的临床意义。
- 结论:与其他模型相比,NMF-MTLR 具有许多优点:卓越的模型辨别力、卓越的校准、有意义的生存时间估计以及每个患者随时间推移的准确生存概率估计。我们主张在临床和研究环境中采用这些癌症生存模型。
微生物组衍生的钴胺素和琥珀酰辅酶A作为改善肛门癌筛查的生物标志物
Microbiome-derived cobalamin and succinyl-CoA as biomarkers for improved screening of anal cancer. Nat Med
- 人乳头瘤病毒可引起侵袭前的高级鳞状上皮内病变 (HSIL),作为肛门生殖器区域癌症的前兆,而微生物组被认为是一个促成因素。感染人类免疫缺陷病毒 (HIV) 的男男性行为者 (MSM) 患肛门癌的风险很高,但目前 HSIL 检测的筛查策略缺乏特异性。
- 在这里,我们研究了肛门微生物组以改善 HSIL 筛查。我们招募了 HIV 感染者,分为发现队列 (n = 167) 和验证队列 (n = 46),他们主要是 (93.9%) 顺性别 MSM,接受高分辨率肛门镜检查和肛门活检进行 HSIL 筛查。
- 我们没有发现与 HSIL 相关的微生物组组成特征,但在两个队列中,产生琥珀酰辅酶 A 和钴胺素的微生物组编码蛋白水平升高与 HSIL 显着相关。在肛门细胞刷中单独测量这些候选生物标志物作为 HSIL 的诊断指标优于肛门细胞学,将敏感性从 91.2% 提高到 96.6%,特异性从 34.1% 提高到 81.8%,并将 82% 的假阳性结果重新分类为真阴性。
- 我们认为这两种微生物组衍生的生物标志物可能会改善当前的肛门癌筛查策略。
其它类
RNA m6A 和 DNA 甲基化之间的串扰调节人多能干细胞中转座元件染色质激活和细胞命运
美国杜克大学医学中心细胞生物学系
- 转座元件 (TE) 是占人类基因组一半以上的寄生 DNA 序列。严格控制 TE 的抑制和激活状态对于基因组完整性、发育、免疫和疾病(包括癌症)至关重要。然而,这一监管具体是如何实现的仍不清楚。
- 在这里,我们开发了一种靶向蛋白质组邻近标记方法来捕获人类胚胎干细胞 (hESC) 中的 TE 相关蛋白。我们发现 RNA N6-甲基腺苷 (m6A) 阅读器 YTHDC2 占据灵长类特异性 TE、LTR7/HERV-H 的基因组位点,特别是通过其与 m6A 修饰的 HERV-H RNA 的相互作用。出乎意料的是,YTHDC2 招募 DNA 5-甲基胞嘧啶 (5mC) 去甲基酶 TET1,从 LTR7/HERV-H 中去除 5mC 并防止表观遗传沉默。从功能上讲,YTHDC2/LTR7 轴抑制 hESC 的神经分化。
- 我们的结果揭示了 RNA m6A 和 DNA 5mC 之间未被充分认识的串扰(真核生物中最丰富的 RNA 和 DNA 调节修饰),以及在 hESC 中这种相互作用控制 TE 活性和细胞命运的事实。
用于监测体内去甲肾上腺素的灵敏多色指示器
Sensitive multicolor indicators for monitoring norepinephrine in vivo. Nat Methods
- 由 G 蛋白偶联受体设计的基因编码指示剂是实现高分辨率体内神经调节剂成像的重要工具。
- 在这里,我们介绍了一系列敏感的多色去甲肾上腺素 (NE) 指示剂,其中包括 nLightG(绿色)和 nLightR(红色)。这些工具报告了体外、离体和体内的内源性 NE 释放,与以前最先进的 GRABNE 指标相比,具有更高的灵敏度、配体选择性和动力学,以及独特的药理学特征。
- 使用 nLightG 的体内多位点光纤光度测量记录,我们可以同时监测小鼠蓝斑和海马中光遗传学诱发的 NE 释放。 nLightG 的双光子成像揭示了海马背侧 CA1 区域的运动和奖励相关的 NE 瞬变。
- 因此,这里介绍的敏感 NE 指标代表了对当前指标库的重要补充,并为彻底调查 NE 系统提供了手段。
Open-3DSIM:开源三维结构照明显微镜重建平台
Open-3DSIM: an open-source three-dimensional structured illumination microscopy reconstruction platform. Nat Methods
中国北京大学未来技术学院生物医学工程系
- Open-3DSIM 是一个用于三维结构照明显微镜的开源重建平台。
- 我们在各种样本和一系列信噪比水平上证明了其相对于其他算法在伪影抑制和高保真度重建方面的优越性能。
- Open-3DSIM 还提供提取偶极子方向的能力,为解释六维 (xyzθλt) 亚细胞结构开辟了新途径。
- 该平台以 MATLAB 代码、Fiji 插件和 Exe 应用程序形式提供,以最大限度地提高用户友好性。
对 5xFAD 小鼠大脑区域的蛋白质组学分析揭示了溶酶体相关蛋白 Arl8b 作为阿尔茨海默病的候选生物标志物
- 阿尔茨海默病(AD)的特点是细胞内和细胞外β-淀粉样蛋白(Aβ)肽的积累。 Aβ 聚集如何扰乱患者和 AD 转基因小鼠模型大脑中的蛋白质组,目前仍不清楚。最先进的质谱 (MS) 方法可以全面检测蛋白质组变化,提供转录组学研究无法获得的相关见解。对 5xFAD 转基因小鼠大脑中渐进性 Aβ 聚集与蛋白质丰度变化之间关系的分析尚未见报道。
- 我们通过免疫组织化学和膜过滤测定定量了 5xFAD 小鼠和对照小鼠海马和皮质中进行性 Aβ 聚集。通过基于 MS 的蛋白质组学使用无标记定量分析不同小鼠组织中的蛋白质变化;使用已建立的管道处理所得的 MS 数据。结果与来自死后 AD 患者大脑的现有蛋白质组数据集进行了对比。最后,使用 ELISA 在 AD 患者和对照的脑脊液 (CSF) 中验证了候选标记物 Arl8b 的丰度变化。
- 实验表明,5xFAD 小鼠海马中 Aβ42 肽的积累速度比皮质中更快,海马中蛋白质丰度变化更大,表明 Aβ42 聚集沉积与大脑区域特异性蛋白质组扰动有关。通过生成时间分辨数据集,我们定义了 Aβ 聚集相关和反相关的蛋白质组变化,其中一部分在 AD 患者死后脑组织中保守,表明 5xFAD 小鼠中的蛋白质组变化模仿了人类 AD 中疾病相关的变化。我们检测到 5xFAD 小鼠海马中 Aβ42 聚集体沉积与溶酶体相关的小 GTP 酶 Arl8b 丰度之间存在正相关性,后者与 5xFAD 大脑中细胞外 Aβ 斑块附近的轴突溶酶体膜一起积累。在人 AD 脑组织中观察到 Arl8b 的异常聚集。 AD患者脑脊液中Arl8b蛋白水平显着升高。
- 我们报告了对 5xFAD 转基因小鼠海马和皮质脑组织的全面生化和蛋白质组学研究,为神经科学界提供了宝贵的资源。我们鉴定出 Arl8b,在 5xFAD 和 AD 患者大脑中存在显着的丰度变化。 Arl8b 可能能够测量 AD 患者进行性溶酶体积累,并作为候选生物标志物具有临床实用性。
经过手术优化的术中释放聚 (I:C) 的水凝胶可预防癌症复发
A surgically optimized intraoperative poly(I:C)-releasing hydrogel prevents cancer recurrence. Cell Rep Med
提有趣的一项研究
- 手术切除原发肿瘤后经常发生复发。在许多癌症中,辅助疗法的疗效有限。手术提供了进入肿瘤微环境的途径,为局部治疗,特别是免疫治疗创造了机会,可以诱导局部和全身的抗癌作用。
- 在这里,我们开发了一种经过手术优化的可生物降解的透明质酸水凝胶,用于在术中持续递送 Toll 样受体 3 激动剂聚 (I:C),并证明它可以在多个小鼠模型中显着减少手术后的肿瘤复发。
- 从机制上讲,Poly(I:C) 可诱导短暂的干扰素 α (IFNα) 反应,通过吸引炎症单核细胞和消耗调节性 T 细胞来重塑肿瘤/伤口微环境。我们证明,预先存在的 IFN 特征可以预测对聚 (I:C) 水凝胶的反应,从而使肿瘤对免疫检查点治疗敏感。一项针对犬软组织肿瘤的兽医试验证实了其安全性、免疫原性和手术可行性。
- 经过手术优化的聚 (I:C) 负载水凝胶提供了一种安全有效的方法来预防癌症复发。
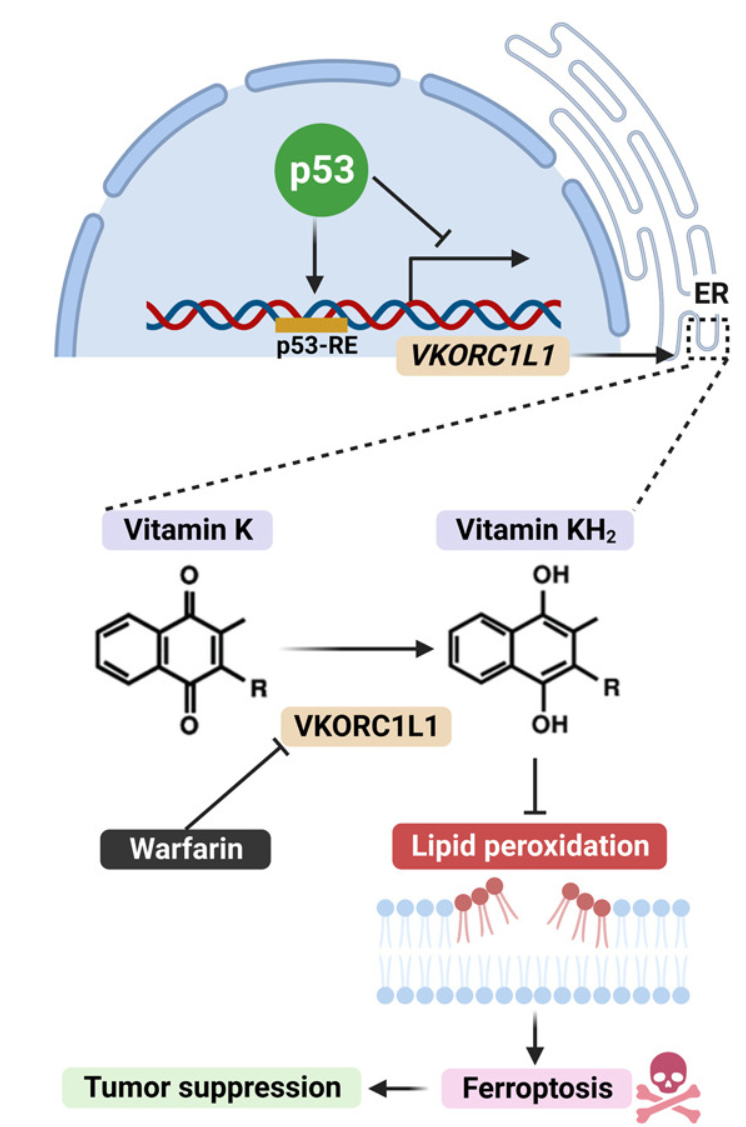
华法林靶向VKORC1L1调节铁死亡治疗癌症
Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab
- 在这里,我们确定维生素 K 环氧化物还原酶复合物亚基 1 like 1 (VKORC1L1) 是一种有效的铁死亡抑制剂。VKORC1L1 通过生成还原型维生素 K对抗磷脂过氧化物,从而保护细胞免遭铁死亡,而与典型的 GSH/GPX4 机制无关。
- 值得注意的是,我们发现 VKORC1L1 也是 p53 的直接转录靶标。 p53 的激活会诱导 VKORC1L1 表达下调,从而使细胞对铁死亡敏感,从而抑制肿瘤。有趣的是,VKORC1L1 的小分子抑制剂华法林 (warfarin) 被广泛用作 FDA 批准的抗凝药物。此外,华法林通过促进免疫缺陷和免疫功能正常的小鼠模型中的铁死亡来抑制肿瘤生长。
- 因此,通过下调 VKORC1L1,p53 通过激活参与维生素 K 代谢的重要铁死亡途径来执行肿瘤抑制功能。我们的研究还表明,华法林是癌症治疗中潜在的再利用药物,特别是对于 VKORC1L1 高表达水平的肿瘤。
早期癌症+化疗幸存者的认知障碍
Early life cancer and chemotherapy lead to cognitive deficits related to alterations in microglial-associated gene expression in prefrontal cortex. Brain Behav Immun
- 罹患白血病后幸存的儿童出现认知困难的风险增加。更好地了解生命早期化疗引起的神经生物学变化将有助于制定治疗策略,以改善白血病幸存者的生活质量。
- 为此,我们使用了翻译相关的小鼠模型,该模型包括将白血病细胞系 (L1210) 注射到出生后第 (P)19 天的小鼠中,然后进行甲氨蝶呤、长春新碱和亚叶酸化疗。化疗结束后一周开始,对雄性和雌性小鼠的社会行为、识别记忆和执行功能(使用5选择串行反应时间任务(5CSRTT)进行测试。在基因表达分析的行为测定结束时收集前额皮质(PFC)和海马体(HPC)。
- 接受生命早期癌症+化疗的小鼠在 5CSRTT 中越来越困难的阶段中进展较慢,并且表现出过早错误的增加,表明冲动行为。研究发现 PFC 中的一组小胶质细胞相关基因与 5CSRTT 的表现和操作反应的获得相关,并且 PFC 和 HPC 中基因表达的长期变化都很明显。
- 这项工作确定了 PFC 和 HPC 中的基因表达变化,这些变化可能是早期接受癌症+化疗的幸存者认知缺陷的基础。
年轻神经胶质祖细胞竞争性地取代成年嵌合小鼠大脑中衰老和患病的人类神经胶质细胞
Young glial progenitor cells competitively replace aged and diseased human glia in the adult chimeric mouse brain. Nat Biotechnol. full html
听上去蛮有趣的研究
- 成人脑细胞之间的竞争尚未得到广泛研究。为了研究健康的神经胶质细胞是否能够在成年前脑中战胜患病的人类神经胶质细胞,我们将由人类胚胎干细胞产生的野生型(WT)人类神经胶质祖细胞(hGPC)移植到已与突变亨廷顿蛋白新生儿嵌合的成年小鼠的纹状体中(mHTT) 表达 hGPC。
- WT hGPC 在竞争中胜出并最终消除了人类亨廷顿病 (HD) 对应物,用健康的神经胶质细胞重新填充了宿主纹状体。单细胞 RNA 测序显示,WT hGPC 在与宿主 HD 胶质细胞相互作用后获得了 YAP1/MYC/E2F 定义的显性竞争表型。 WT hGPC 还击败了新生儿移植的较老驻留同基因 WT 细胞,这表明竞争成功主要取决于竞争群体的相对年龄,而不是 mHTT 的存在。
- 这些数据表明,成年大脑中衰老和患病的人类神经胶质细胞可能被年轻的健康 hGPC 广泛取代,这提出了替代衰老和患病人类神经胶质细胞的治疗策略。
通过基于 CRISPR 的条形码对发育中的小鼠大脑中的单细胞谱系进行全面的时空图谱
Comprehensive spatiotemporal mapping of single-cell lineages in developing mouse brain by CRISPR-based barcoding. Nat Methods
中国科学院神经科学研究所
- 发育神经科学的一个根本兴趣在于绘制大脑内完整单细胞谱系的能力。为此,我们开发了一种基于 CRISPR 编辑的谱系特异性追踪 (CREST) 方法,用于 Cre 小鼠的克隆追踪。然后,我们使用两种基于 CREST 的互补策略来绘制发育中的小鼠腹侧中脑 (vMB) 的单细胞谱系。
- 通过应用快照CREST (snapCREST),我们构建了正在发育的vMB的时空谱系景观,并鉴定了六种祖细胞原型,它们可以代表单个vMB祖细胞的主要克隆命运,以及底板中指定谷氨酸能、多巴胺能或两种神经元的三种不同的克隆谱系。我们进一步创建了 pandaCREST(祖细胞和衍生物关联 CREST),将体内祖细胞的转录组与其分化潜力相关联。我们鉴定了多巴胺能神经元的多个起源,并证明转录组定义的祖细胞类型包含异质祖细胞,每个祖细胞具有不同的克隆命运和分子特征。因此,CREST 方法和策略允许进行全面的单细胞谱系分析,可以为神经规范背后的分子程序提供新的见解。
SARS-CoV-2 特异性 T 细胞治疗重症 COVID-19:一项随机 1/2 期试验
SARS-CoV-2-specific T cell therapy for severe COVID-19: a randomized phase 1/2 trial. Nat Med
- 尽管取得了进展,但很少有治疗方法对 2019 年严重冠状病毒病 (COVID-19) 表现出疗效。在不同的背景下,病毒特异性 T 细胞已被证明是安全有效的。
- 我们进行了一项随机 (2:1)、开放标签、1/2 期试验,以评估现成的、部分人类白细胞抗原 (HLA) 匹配、恢复期供体来源的严重急性呼吸综合征的安全性和有效性与 Delta 变异占主导地位期间的 SoC 相比,冠状病毒 2 (SARS-CoV-2) 特异性 T 细胞 (CoV-2-ST) 与重症 COVID-19 患者的标准护理 (SoC) 相结合。经过剂量递增的 1 期安全性研究后,90 名参与者被随机分配接受 CoV-2-ST+SoC (n = 60) 或仅接受 SoC (n = 30)。该研究的共同主要目标是综合考虑恢复时间和 30 天恢复率,以及接受 CoV-2-ST+SoC 而非 SoC 的患者体内 CoV-2-ST 的扩增。关键的次要目标是第 60 天的存活率。
- CoV-2-ST+SoC 治疗安全且耐受性良好。该研究达到了主要复合终点(CoV-2-ST+SoC 与 SoC:恢复率分别为 65% 与 38%,P = 0.017;中位恢复时间分别为 11 天与未达到,P = 0.052;恢复率比 1.71 (95% 置信区间 1.03-2.83,P = 0.036))以及与 SoC 相比 CoV-2-ST 显着扩展的共同主要目标(CoV-2-ST+SoC 与 SoC,P = 0.047)。
- 总体而言,对于住院的重症 COVID-19 患者,使用 CoV-2-ST 进行过继免疫治疗是可行且安全的。需要更大规模的试验来加强对重症 COVID-19 的临床益处的初步证据。
机器学习/组学类
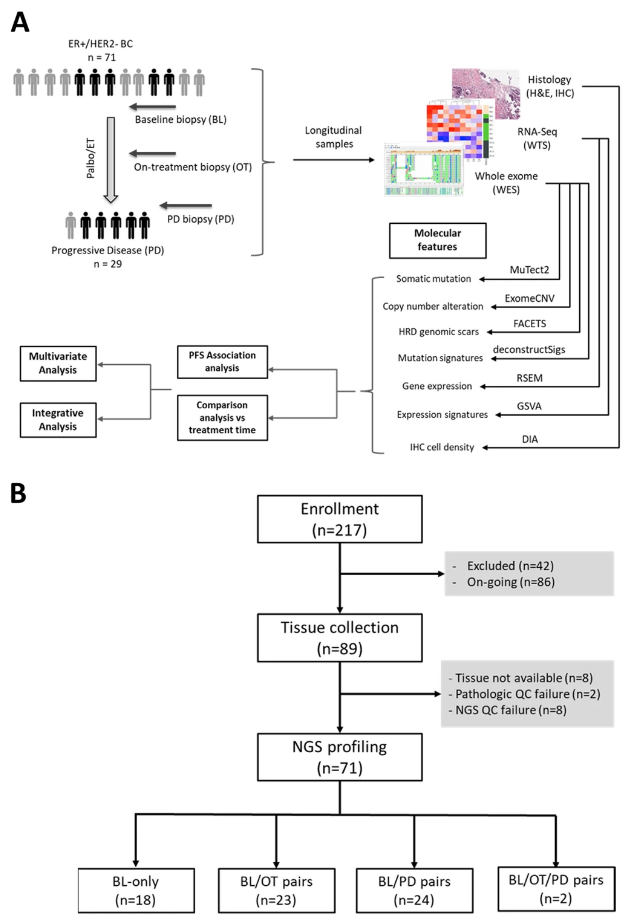
HR 阳性/HER2 阴性转移性乳腺癌帕博西尼耐药的纵向多组学研究
- 细胞周期蛋白依赖性激酶4/6抑制剂(CDK4/6)联合内分泌治疗(ET)是治疗激素受体阳性/人表皮受体2阴性转移性乳腺癌(HR+/HER2-MBC)患者的有效治疗方法;然而,抵抗现象很常见,而且人们对此知之甚少。对接受哌柏西利加 ET 的患者治疗前和治疗后的肿瘤进行了全面的基因组和转录组分析,以描绘耐药性的分子机制。
- 收集 2017 年至 2020 年在三星医疗中心和首尔国立大学医院接受哌柏西利加芳香酶抑制剂或氟维司群治疗的 89 名 HR+/HER2- MBC 患者(包括复发和/或转移性疾病患者)的组织。对治疗前、治疗中(6 周和/或 12 周)和进展后获得的肿瘤活检和血液样本进行 RNA 测序和全外显子组测序。进行 Cox 回归分析以确定与无进展生存期相关的临床和基因组变量。
- 鉴定出与不良预后相关的新标志物,包括同源修复缺陷(HRD)引起的基因组疤痕特征、雌激素反应特征以及具有不同分子特征的四个预后簇。具有 TP53 突变且与独特的 HRD 高簇共存的肿瘤对 Palbociclib 加 ET 反应较差。配对治疗前和治疗后样本的比较显示,肿瘤中 APOBEC 突变特征变得丰富,并且许多肿瘤转变为具有雌激素独立特征的侵袭性分子亚型。我们在 RB1、ESR1、PTEN 和 KMT2C 中发现了疾病进展时频繁发生的基因组改变。
- 我们确定了与不良预后相关的新分子特征以及可用于克服 CKD4/6 加 ET 耐药性的分子机制。
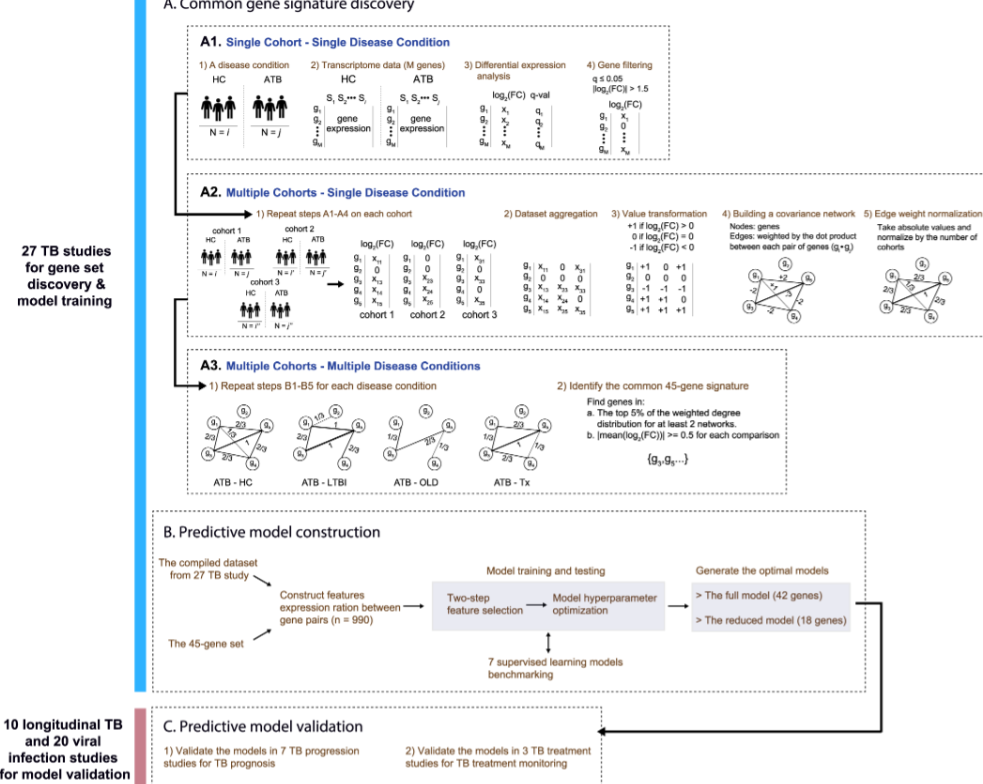
针对结核病预后和治疗反应的不同临床队列的基因特征发现和系统验证
Gene signature discovery and systematic validation across diverse clinical cohorts for TB prognosis and response to treatment. PLoS Comput Biol
- 虽然血液基因特征在结核病 (TB) 诊断和治疗监测中显示出希望,但来自单个队列的大多数特征可能不足以捕获人群和个体中的结核病异质性。
- 在这里,我们报告了一种新的通用方法,将基于网络的元分析与机器学习建模相结合,以利用研究之间异质性的力量。来自 57 项研究(37 项结核病和 20 项病毒感染)的转录组数据集涵盖人口统计和结核病状态,用于基因特征发现以及模型训练和验证。
- 基于网络的荟萃分析确定了跨研究的活动性结核病特有的共同 45 个基因特征。然后使用全部或部分 45 个基因特征建立两个优化的随机森林回归模型,以模拟从结核分枝杆菌感染到疾病和治疗反应的连续体。
- 在模型验证中,使用汇集的多队列数据集来模拟现实世界的环境,该模型为 2.5 年期间的初期活动性结核病风险提供了稳健的预测性能,AUROC 为 0.85、74.2% 的敏感性和 78.3% 的特异性,接近 WHO 目标产品概况中预测结核病进展的最低标准(>75% 的敏感性和 >75% 的特异性)。此外,该模型强烈区分活动性结核病和病毒感染(AUROC 0.93,95% CI 0.91-0.94)。对于治疗监测,该模型生成的结核病评分与一段时间内的治疗反应在统计上相关,并且甚至在治疗开始之前就可以预测标准治疗的临床结果。
- 我们展示了一种端到端基因特征模型开发方案,该方案考虑了结核病风险评估和治疗监测的异质性。
DREAM:人类复杂疾病成药性评估
DREAM: an R package for druggability evaluation of human complex diseases. Bioinformatics
- 从头药物开发是一个漫长而昂贵的过程,从设计到临床前测试都面临着巨大的挑战,使得进入市场缓慢而困难。这一限制为药物再利用的发展铺平了道路。药物再利用包括重复使用已批准的药物,这些药物是为其他治疗适应症而开发的。尽管在过去十年中为了实现临床相关的药物再利用预测已经进行了一些努力,但在实际药理学治疗中使用的再利用药物的数量仍然有限。一方面,机械方法,包括基于概况和基于网络的方法,利用有关药物敏感性和扰动概况以及疾病转录组学概况的大量数据。另一方面,以化学为中心的方法,包括基于结构的方法,考虑了药物及其分子靶标的内在结构特性。机械方法和以化学为中心的方法之间的整合不佳是药物再利用预测难以转化为临床的主要限制因素之一。
- 我们引入了 DREAM,这是一个 R 包,旨在将机械和化学中心方法集成到统一的计算工作流程中。 DREAM 致力于利用可靠的药物再利用预测对感兴趣的病理状况进行成药性评估。此外,用户还可以得出推定适合联合治疗的优化药物组。为了展示 DREAM 软件包的功能,我们报告了一个关于特应性皮炎的案例研究。
- 可用性:DREAM 可在 https://github.com/fhaive/dream 免费获取。 DREAM 的 docker 镜像位于:https://hub.docker.com/r/fhaive/dream。
GradPose:基于梯度下降的快速蛋白质结构叠加
- 分子动力学 (MD) 和对接等计算模拟为蛋白质的动力学和相互作用构象提供了重要的见解,补充了确定蛋白质结构的实验方法。这些方法通常会生成数百万个蛋白质构象,需要高效的结构比较和聚类方法来分析结果。
- 在本文中,我们介绍了 GradPose,一种快速且内存高效的结构叠加工具,用于这些大规模模拟生成的模型。 GradPose 使用梯度下降通过优化旋转四元数来最佳地叠加结构,并且与参考结构相比可以处理插入和删除。它能够在标准硬件上叠加数千到数百万个蛋白质结构,并利用多个 CPU 核心和 CUDA 加速(如果可用)来进一步减少叠加时间。
- 我们的结果表明,GradPose 总体上优于传统方法,速度提高了 2 至 65 倍,内存需求减少了 1.7 至 48 倍,较大的蛋白质结构受益最多。我们观察到,传统方法仅在处理由 ∼20 个残基组成的非常小的蛋白质时优于 GradPose。 GradPose的前提是残基-残基对应关系是预先确定的。通过 GradPose,我们的目标是提供一种计算高效的解决方案,以应对计算模拟领域中有效处理结构对齐需求的挑战。
- 源代码可在 https://github.com/X-lab-3D/GradPose 免费获取; DOI:10.5281/zenodo.7671922。
AGHmatrix:遗传关系矩阵
AGHmatrix: genetic relationship matrices in R. Bioinformatics
- 根据谱系和基因组数据计算出的亲属之间的相似性对于遗传学家和生态学家来说是一个重要资源,他们有兴趣了解基因如何影响表型变异、适应性适应和种群动态。
- AGHmatrix 软件是一个 R 软件包,专注于构建谱系(A 矩阵)和/或分子标记(G 矩阵),并有可能构建由分子标记校正的谱系组合矩阵(H 矩阵)。该软件旨在估计任何倍性水平的关系,还包括与过滤分子标记、计算连锁不平衡 (LD) 和检查大数据集中的谱系错误相关的辅助功能。计算关系矩阵后,AGH 矩阵的结果可用于不同的环境,包括预测(基因组)估计育种值和全基因组关联研究 (GWAS)。
- 可用性:AGHmatrix v2.1.0 可在 CRAN 中的 GPL-3 许可下获取(网址为 https://cran.r-project.org/web/packages/AGHmatrix/index.html),也可在 GitHub 中获取(网址为 https://github.com/rramadeu/AGHmatrix)。它有一个全面的教程,并附有真实的数据示例。
电压测序:全光学突触后连接组引导的单细胞转录组学
Voltage-Seq: all-optical postsynaptic connectome-guided single-cell transcriptomics. Nat Methods. full html
- 了解神经元信息的路由需要连接的功能特征。神经元投射招募具有不同突触后反应类型(PRT)的大型突触后整体。 PRT 通常通过低通量全细胞电生理学进行探测,并不是单细胞 RNA 测序 (scRNA-seq) 的选择标准。
- 为了克服这些限制并基于特定的 PRT 来靶向神经元以进行体细胞收获和随后的 scRNA 测序,我们创建了电压测序。我们建立了全光电压成像,并记录了由腹内侧下丘脑 (VMH) 末端的光遗传学激活引起的小鼠导水管周围灰质 (PAG) 中 8,347 个神经元的 PRT。 PRT 在整个 VMH-PAG 连接组中进行分类和空间解析。
- 我们构建了一个名为 VoltView 的现场分析工具,在使用 VMH-PAG 连接组数据库作为参考的分类器的指导下,将体细胞采集导航至目标 PRT。我们通过在 PAG 中定位 VMH 驱动的 γ-氨基丁酸能神经元来演示电压序列,仅由 VoltView 中的现场分类指导。
L-GIREMI 在长读长 RNA-seq 中发现 RNA 编辑位点
L-GIREMI uncovers RNA editing sites in long-read RNA-seq. Genome Biol
美国加州大学综合生物学和生理学系
- 尽管长读长 RNA-seq 越来越多地应用于表征全长转录本,但它也可以检测核苷酸变异,例如基因突变或 RNA 编辑位点,而这方面的研究还远远不够。
- 在这里,我们提出了一项深入研究来检测和分析长读长 RNA-seq 中的 RNA 编辑位点。我们的新方法 L-GIREMI 可有效处理测序错误和读取偏差。 L-GIREMI 应用于 PacBio RNA-seq 数据,可提供高精度的 RNA 编辑识别。此外,我们的分析揭示了有关单分子和双链 RNA 结构中 RNA 编辑发生的新见解。
- L-GIREMI 为研究长读长 RNA 序列中的核苷酸变异提供了一种有价值的方法。
UNMF:多维组学数据的统一非负矩阵分解
UNMF: a unified nonnegative matrix factorization for multi-dimensional omics data. Brief Bioinform. full pdf & supplementary materials
日本名古屋大学医学研究生院系统生物学系
非负矩阵分解(NMF)是一种非常常见的方法。对GSClassifier设计可能有提示作用。
- 因子分析,从主成分分析到非负矩阵分解,代表了分析多维数据以提取有价值模式的最重要方法,并且越来越多地应用于以张量形式表示的多维组学数据集的背景下。然而,传统的分析方法严重依赖于数据本身的格式和结构,即使这些变化很小,分析人员也必须改变数据分析策略和技术,并花费大量时间进行数据预处理。此外,在数据中存在缺失值的情况下,许多传统方法无法按原样应用。
- 我们提出了一个新的统计框架,即统一非负矩阵分解(UNMF),用于在混乱的生物数据集中查找信息模式。 UNMF 旨在整洁的数据格式和结构,使数据分析更加容易,并简化数据分析工具的开发。 UNMF 可以处理各种数据结构和格式,并与张量数据无缝协作,包括缺失观测值和重复测量值。
- UNMF 的实用性通过其对多个多维组学数据的应用得到证明,为分析和集成提供用户友好且统一的功能。它的应用对生命科学界具有巨大的潜力。
HiC4D:使用残差 ConvLSTM 预测时空 Hi-C 数据
HiC4D: forecasting spatiotemporal Hi-C data with residual ConvLSTM. Brief Bioinform
- Hi-C 实验已广泛用于基因组结构的研究。在过去的几年中,时空 Hi-C 在很大程度上促进了基因组动态重组的研究。然而,时空 Hi-C 数据的计算建模和预测尚未见于文献。
- 我们提出 HiC4D 来处理预测时空 Hi-C 数据的问题。我们设计并测试了一种新颖的网络,并将其命名为残差ConvLSTM(ResConvLSTM),它是残差网络和卷积长短期记忆(ConvLSTM)的组合。
- 我们评估了新的 ResConvLSTM 网络,并将其与其他五种方法进行了比较,包括我们设计作为基线方法的朴素网络 (NaiveNet) 和文献中的四种出色的视频预测方法:ConvLSTM、时空 LSTM (ST-LSTM)、自注意力 LSTM (SA-LSTM) 和简单视频预测 (SimVP)。我们使用八个不同的时空 Hi-C 数据集进行盲测,其中两个来自小鼠胚胎发生,一个来自体细胞核移植(SCNT)胚胎,三个来自不同物种的胚胎发生数据集和两个非胚胎发生数据集。
- 我们的评估结果表明,在准确预测未来时间步长的 Hi-C 接触矩阵方面,我们的 ResConvLSTM 网络在八个盲测数据集上几乎总是优于其他方法。我们的基准测试还表明,我们基准测试的所有方法都可以成功恢复实验 Hi-C 接触矩阵所调用的拓扑关联域的边界。
- 综上所述,我们的基准测试表明 HiC4D 是预测时空 Hi-C 数据的有效工具。 HiC4D 可在 http://dna.cs.miami.edu/HiC4D/ 和 https://github.com/zwang-bioinformatics/HiC4D/ 上公开获取。
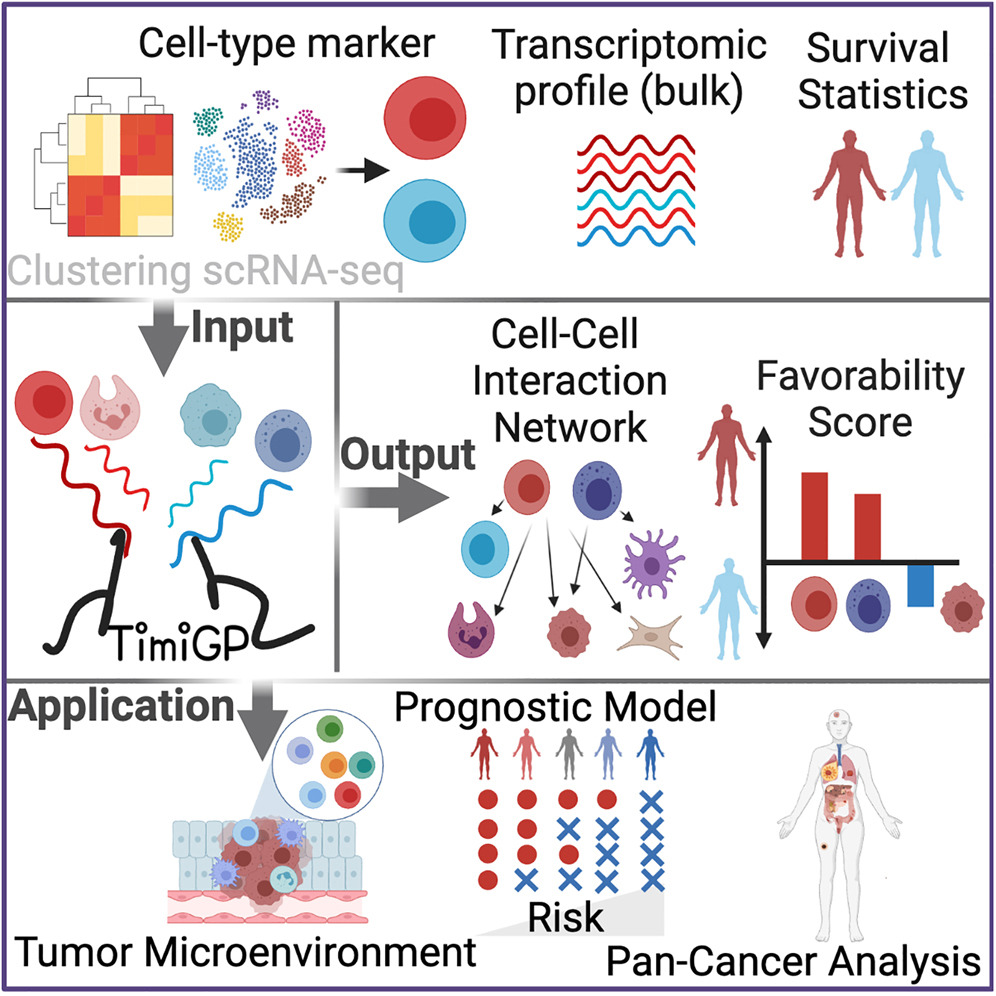
(~ ̄▽ ̄)~ TimiGP:通过基因对推断肿瘤免疫微环境中的细胞间相互作用和预后关联
TimiGP: Inferring cell-cell interactions and prognostic associations in the tumor immune microenvironment through gene pairs. Cell Rep Med. full html; full pdf
德克萨斯大学 MD 安德森癌症中心基因组医学系
与GSClassifier-PAD高度相似,但研究背景中并没有引用 (ฅ´ω`ฅ)
- 确定肿瘤微环境中不同免疫细胞类型的预后关联对于了解癌症生物学和开发新的治疗策略至关重要。然而,这在某些癌症类型中具有挑战性,因为不同免疫亚群的丰度高度相关。
- 在本研究中,我们开发了一种名为 TimiGP 的计算方法来克服这一挑战。基于大量基因表达和存活数据,TimiGP 推断细胞间相互作用,揭示免疫细胞相对丰度与预后之间的关联。正如在转移性黑色素瘤中所证明的那样,TimiGP 根据已识别的细胞间相互作用优先考虑对预后至关重要的免疫细胞。当应用于七个独立的黑色素瘤数据集并且使用不同的细胞类型标记集作为输入时,TimiGP 获得了高度一致的结果。
- 此外,TimiGP 可以利用单细胞 RNA 测序数据以高分辨率描绘多种癌症类型的肿瘤免疫微环境。
跨物种代谢网络分析
Inferring and comparing metabolism across heterogeneous sets of annotated genomes using AuCoMe. Genome Res
- 基因组规模代谢网络(genome-scale metabolic networks, GSMN)的比较分析可能会产生有关物种生物学、进化和适应的重要信息。然而,它受到结构和功能基因组注释的质量和完整性的高度异质性的阻碍,这可能会使这种比较的结果产生偏差。
- 为了解决这个问题,我们开发了 AuCoMe,这是一个管道,可以从一组异构的注释基因组中自动重建同质 GSMN,而无需丢弃可用的手动注释。
- 我们使用三个数据集(一种细菌、一种真菌和一种藻类)测试了 AuCoMe,结果表明它成功减少了技术偏差,同时捕获了每种生物体的代谢特异性。我们的结果还指出了进化距离较远的藻类之间共有和不同的代谢特征,强调了 AuCoMe 加速整个生命树代谢进化的广泛探索的潜力。
CoDoC流程与AI-临床医生共决策
Google DeepMind
- 基于深度学习的预测人工智能(AI)系统已被证明可以在多种医学成像环境中实现专家级的疾病识别,但在临床医生准确诊断的情况下可能会出错,反之亦然。
- 我们开发了互补驱动的延期临床工作流程 (CoDoC),这是一个可以学习在预测 AI 模型的意见和临床工作流程之间做出决定的系统。在筛查乳腺癌或结核病 (TB) 的临床工作流程中,CoDoC 相对于仅临床医生或仅 AI 基线提高了准确性。
- 对于乳腺癌筛查,与英国筛查项目中的双读仲裁相比,CoDoC 在相同假阴性率的情况下将假阳性减少了 25%,同时临床医生的工作量减少了 66%。对于结核病分类,与独立的人工智能和临床工作流程相比,CoDoC 在五个商用预测人工智能系统中的三个系统中,在相同的假阴性率下,假阳性率降低了 5-15%。为了促进 CoDoC 在新颖的未来临床环境中的部署,
- 我们提出的结果表明 CoDoC 的性能增益在多个变化轴(成像模式、临床环境和预测人工智能系统)上得以持续,并讨论了我们评估的局限性以及进一步验证的领域被需要。我们提供开源实现以鼓励进一步的研究和应用。
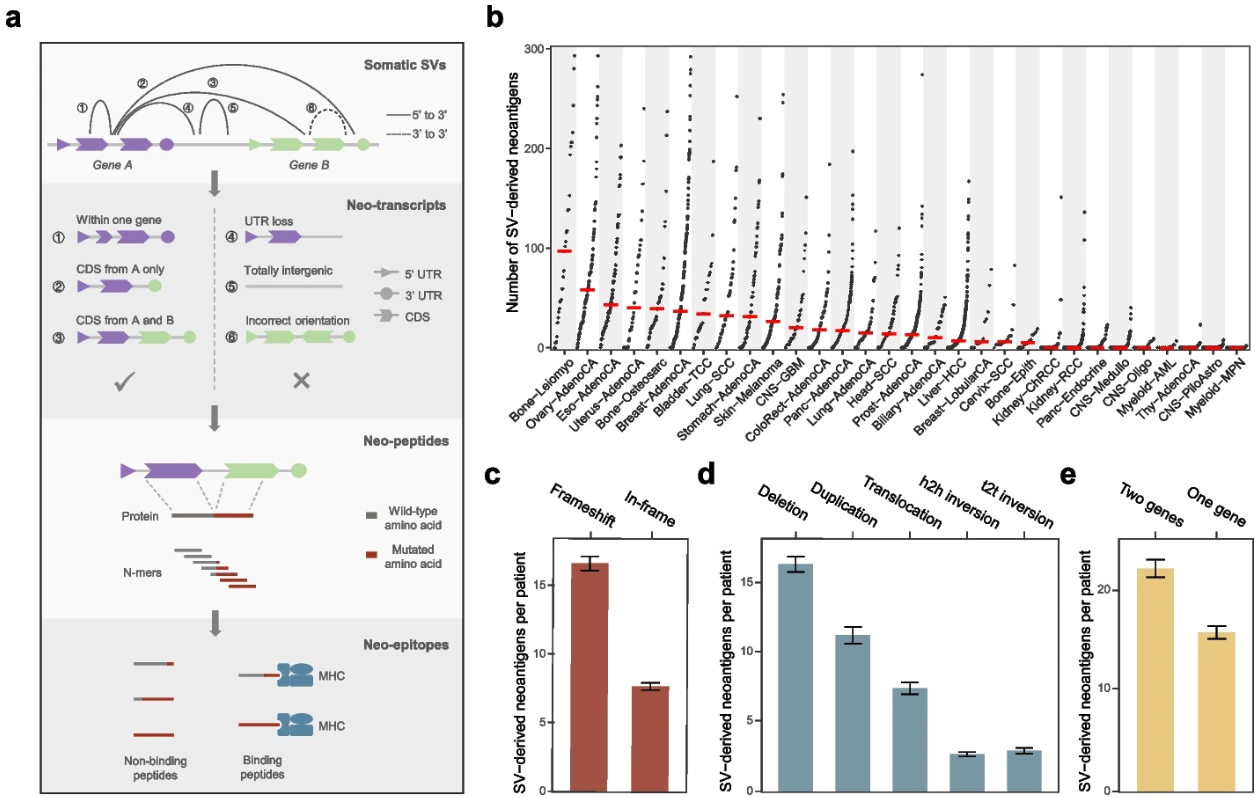
结构变异相关新抗原的泛癌分析
Comprehensive analysis of neoantigens derived from structural variation across whole genomes from 2528 tumors. Genome Biol
北京大学数学科学学院
- 新抗原对于抗肿瘤免疫至关重要,长期以来一直被视为有希望的治疗靶点。然而,目前的新抗原分析主要集中在单核苷酸变异(SNV)和插入缺失突变上,很少考虑在癌症中也普遍存在的结构变异(SV)。
- 在这里,我们开发了一种称为 NeoSV 的计算方法,它将 SV 注释、蛋白质片段化和 MHC 结合预测结合在一起,以预测 SV 衍生的新抗原。
- 对 2528 个全基因组的分析表明,SV 在数量和质量上对新抗原库都有显着贡献。虽然大多数新抗原是患者特异性的,但在乳腺癌、卵巢癌和胃肠道癌中,共享新抗原的发生率很高。我们观察到对 SV 衍生新抗原的广泛免疫编辑,尤其是克隆事件,这表明它们的免疫原性潜力。我们还证明,基因组改变相关的新抗原负荷整合了SV衍生的新抗原,比肿瘤新抗原负荷更好地描述了肿瘤免疫相互作用,并且可以改善患者对免疫治疗的选择。
- 我们的研究填补了当前新抗原库的空白,并为癌症疫苗的开发提供了宝贵的资源。
MAPT 致病性变异外周血细胞的scRNA-Seq研究
- 背景:来自小鼠模型的新证据开始阐明大脑对 tau 病理学的免疫反应,但人们对人类这种反应的性质知之甚少。此外,目前尚不清楚 tau 蛋白病理学和大脑内局部炎症反应在多大程度上影响更广泛的免疫系统。
- 方法:为了解决这些问题,我们对 MAPT 致病性变异携带者(编码 tau 的基因 (n = 8))和健康非携带者的外周血单核细胞 (PBMC) 进行了单细胞 RNA 测序 (scRNA-seq)。控制(n = 8)。我们的 scRNA-seq 分析的主要结果通过流式细胞术、液滴数字 (dd)PCR 和公开转录组数据集的二次分析得到了证实和扩展。
- 结果:对约 181,000 个个体 PBMC 转录组的分析表明,MAPT 致病性变异携带者的单核细胞和自然杀伤 (NK) 细胞存在显着差异表达。特别是,我们观察到单核细胞和 NK 细胞中 CX3CR1 的表达显着减少,CX3CR1 是编码 fractalkine 受体的基因,已知该基因可调节小鼠模型中的 tau 病理学。我们还观察到非经典单核细胞的丰度显着减少,并且非经典单核细胞标记基因(包括 FCGR3A)的表达失调。最后,我们发现了 TMEM176A 和 TMEM176B 的减少,这两个基因被认为与人类小胶质细胞的炎症反应有关,但在外周单核细胞中的功能尚不清楚。我们通过流式细胞术证实了非经典单核细胞的减少,并使用 ddPCR 在我们的 scRNA-seq 数据中证实了选定的生物学相关基因的差异表达。
- 结论:我们的结果表明,人外周免疫细胞的表达和丰度受到 tau 相关病理生理变化的调节。 CX3CR1 和非经典单核细胞尤其将成为未来工作的重点,探索这些外周信号在其他 tau 相关神经退行性疾病中的作用。
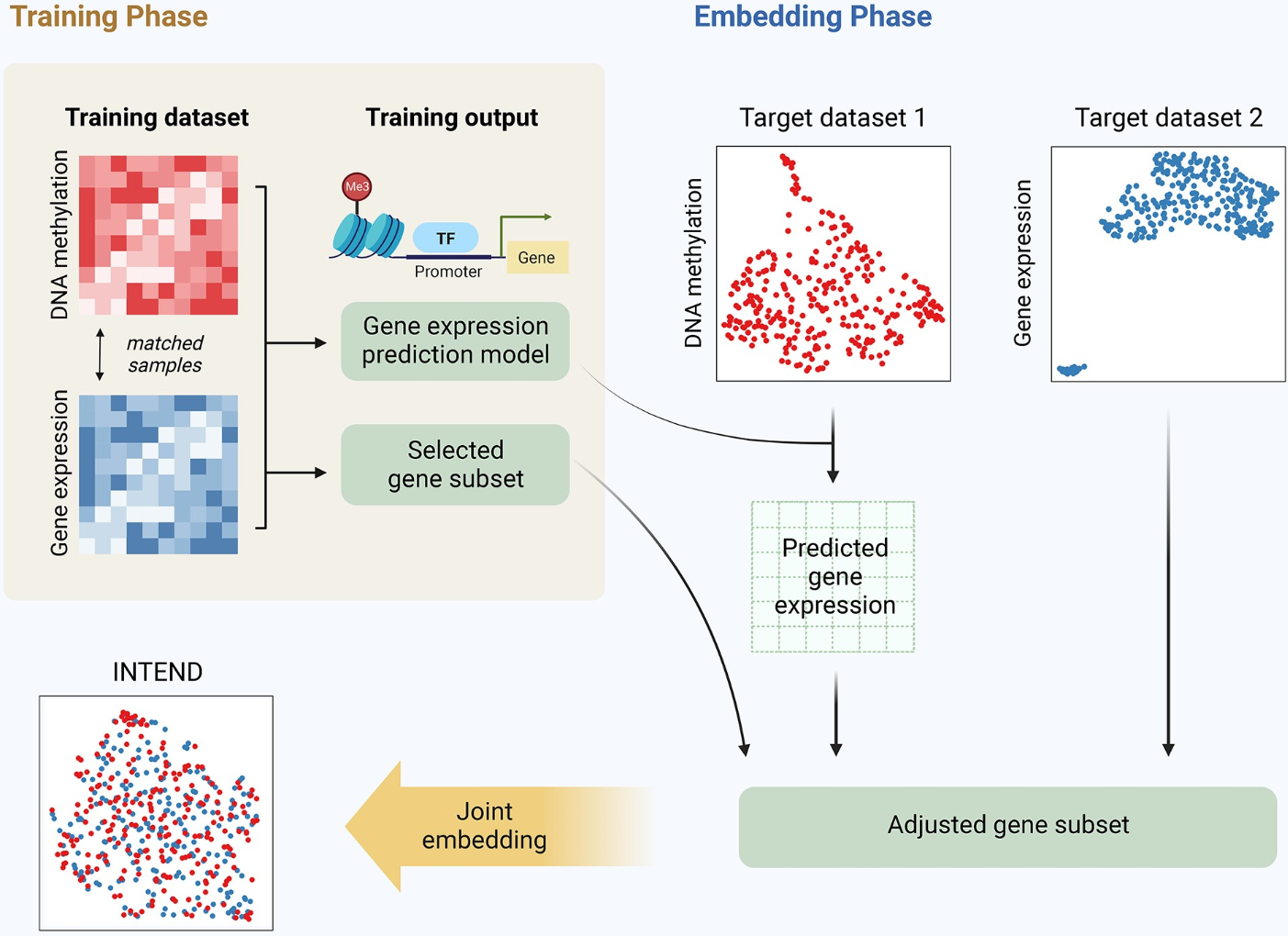
(~ ̄▽ ̄)~ INTEND集成涵盖不相交样本集的基因表达和 DNA 甲基化数据集
Integration of gene expression and DNA methylation data across different experiments. Nucleic Acids Res
很重要,可能与GSClassifier有关。
- 多组学数据集的综合分析已被证明在癌症研究和精准医学中极其有价值。然而,从相同样本中获取多模态数据通常很困难。整合不同组学的多个数据集仍然是一个挑战,只有少数可用的算法被开发来解决它。
- 在这里,我们提出了 INTEND(转录组学和表观组学数据的集成),这是一种新的算法,用于集成涵盖不相交样本集的基因表达和 DNA 甲基化数据集。为了实现集成,INTEND 通过对同一组样本测量的多组学数据进行训练来学习两个组学之间的预测模型。
- 在对涵盖 4329 名患者的 11 个 TCGA(癌症基因组图谱)癌症数据集进行的综合测试中,与四种最先进的集成算法相比,INTEND 取得了显着优越的结果。我们还证明了 INTEND 在对来自不同来源的两个肺腺癌单组学数据集进行联合分析时揭示 DNA 甲基化与基因表达调控之间联系的能力。 INTEND 的数据驱动方法使其成为有价值的多组学数据集成工具。
- INTEND 的代码可在 https://github.com/Shamir-Lab/INTEND 获取。
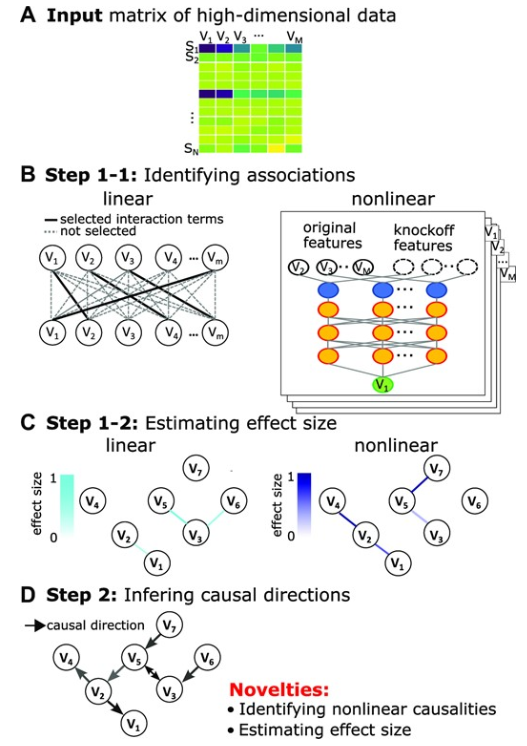
DAG-deepVASE识别生物数据中的非线性因果关系
- 背景:了解因果结构有助于识别复杂疾病的危险因素、疾病机制和候选疗法。然而,尽管复杂的生物系统具有非线性关联的特点,但现有的因果推理生物信息学方法无法识别非线性关系并估计其效应大小。
- 结果:为了克服这些限制,我们开发了第一种计算方法,该方法使用深度神经网络方法与仿冒框架相结合,显式地学习非线性因果关系并估计效应大小,称为使用深度学习变量选择的因果有向无环图(DAG-deepVASE)。
- 使用不同场景的模拟数据并识别各种疾病的分子和临床数据中已知和新颖的因果关系,我们证明 DAG-deepVASE 在识别真实和已知因果关系方面始终优于现有方法。在分析中,我们还说明了识别非线性因果关系并估计其效应大小如何帮助理解复杂的疾病病理学,这是使用其他方法不可能实现的。
- 结论:凭借这些优势,DAG-deepVASE 的应用可以帮助在生物医学研究和临床试验中识别驱动基因和治疗药物。
在资源有限的设备上进行高效的实时选择性基因组测序
Efficient real-time selective genome sequencing on resource-constrained devices. Gigascience
- 背景:第三代纳米孔测序仪提供选择性测序或“Read Until”功能,允许实时分析基因组读数,如果不属于“感兴趣”的基因组区域,则可以中途放弃。这种选择性测序为快速、低成本基因测试等重要应用打开了大门。为了使选择性测序有效,分析的延迟应尽可能低,以便尽早拒绝不必要的读取。然而,针对此问题采用子序列动态时间规整 (sDTW) 算法的现有方法计算量太大,以至于具有数十个 CPU 核心的大型工作站仍然难以跟上手机大小的 MinION 定序器的数据速率。
- 结果:在本文中,我们提出了硬件加速读取(HARU),这是一种基于资源高效的硬件软件协同设计的方法,该方法利用具有片上现场可编程功能的低成本、便携式异构多处理器片上系统平台门阵列 (FPGA) 来加速基于 sDTW 的 Read Until 算法。实验结果表明,嵌入 4 核 ARM 处理器的 Xilinx FPGA 上的 HARU 比在具有 36 个处理器的复杂服务器上运行的高度优化的多线程软件版本快约 2.5 倍(比现有未优化的多线程软件快约 85 倍)。用于 SARS-CoV-2 数据集的英特尔至强处理器核心。 HARU 的能耗比在 36 核服务器上执行的相同应用程序低 2 个数量级。
- 结论:HARU 证明,通过严格的硬件软件优化,可以在资源受限的设备上进行纳米孔选择性测序。 HARU sDTW 模块的源代码可在 https://github.com/beebdev/HARU 上以开源形式获取,使用 HARU 的示例应用程序位于 https://github.com/beebdev/sigfish-haru。
Python版WGCNA
PyWGCNA: A Python package for weighted gene co-expression network analysis. Bioinformatics
欢迎欢迎!
- 加权基因共表达网络分析 (WGCNA) 经常用于识别在许多 RNA-seq 样本中共表达的基因模块。然而,当前的 R 实现速度很慢,并非旨在比较多个 WGCNA 网络之间的模块,并且其结果可能难以解释和可视化。
- 我们引入了 PyWGCNA Python 包,该包旨在从大型 RNA-seq 数据集中识别共表达模块。 PyWGCNA 比 R 版本的 WGCNA 具有更快的实现速度,并具有几个附加的下游分析模块,用于使用 GO、KEGG 和 REACTOME 进行功能富集分析、蛋白质-蛋白质相互作用的模块间分析以及多个共表达模块与每个模块的比较其他和/或外部基因列表,例如来自单细胞的标记基因。
- 我们将 PyWGCNA 应用于来自 MODEL-AD 的两个不同的大批量 RNA-seq 数据集,以识别与基因型相关的模块。我们将生成的模块相互比较,以找到在数据集中具有显着重叠的模块形式的共享共表达签名。
- 可用性:Python 3 的 PyWGCNA 库可在 PyPi 上获取(网址为 pypi.org/project/PyWGCNA),也可在 GitHub 上获取(网址为 github.com/mortazavilab/PyWGCNA)。
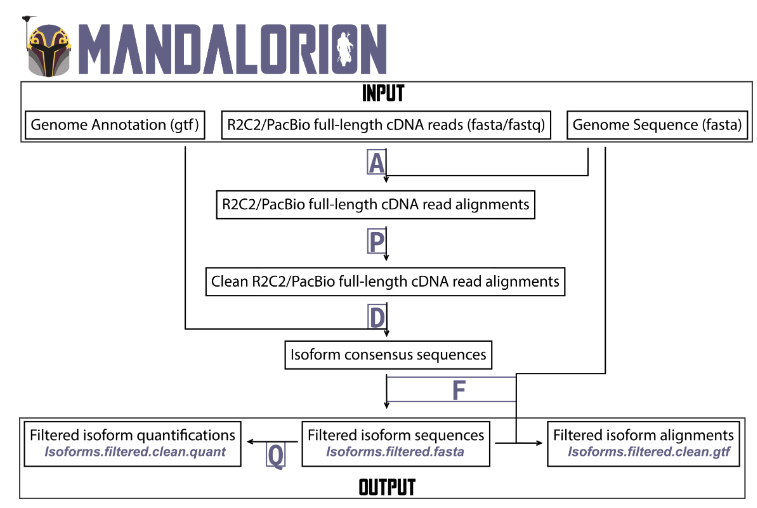
Mandalorion:从RNA-Seq中识别异构体
Identifying and quantifying isoforms from accurate full-length transcriptome sequencing reads with Mandalorion. Genome Biol
- 在这篇手稿中,我们介绍并基准 Mandalorion v4.1,用于全长转录组测序读数的识别和量化。它进一步改进了 LRGASP 联盟挑战赛 中使用的 Mandalorion v3.6 本来就很强大的性能。
- 通过处理真实和模拟数据,我们展示了 Mandalorion 的三个主要特征:首先,基于 Mandalorion 的亚型识别具有非常高的精度,即使在没有任何基因组注释的情况下也能保持高召回率。其次,Mandalorion 量化的同种型读取计数显示与模拟读取计数具有高度相关性。第三,Mandalorion 鉴定的亚型密切反映了它们所基于的全长转录组测序数据集。
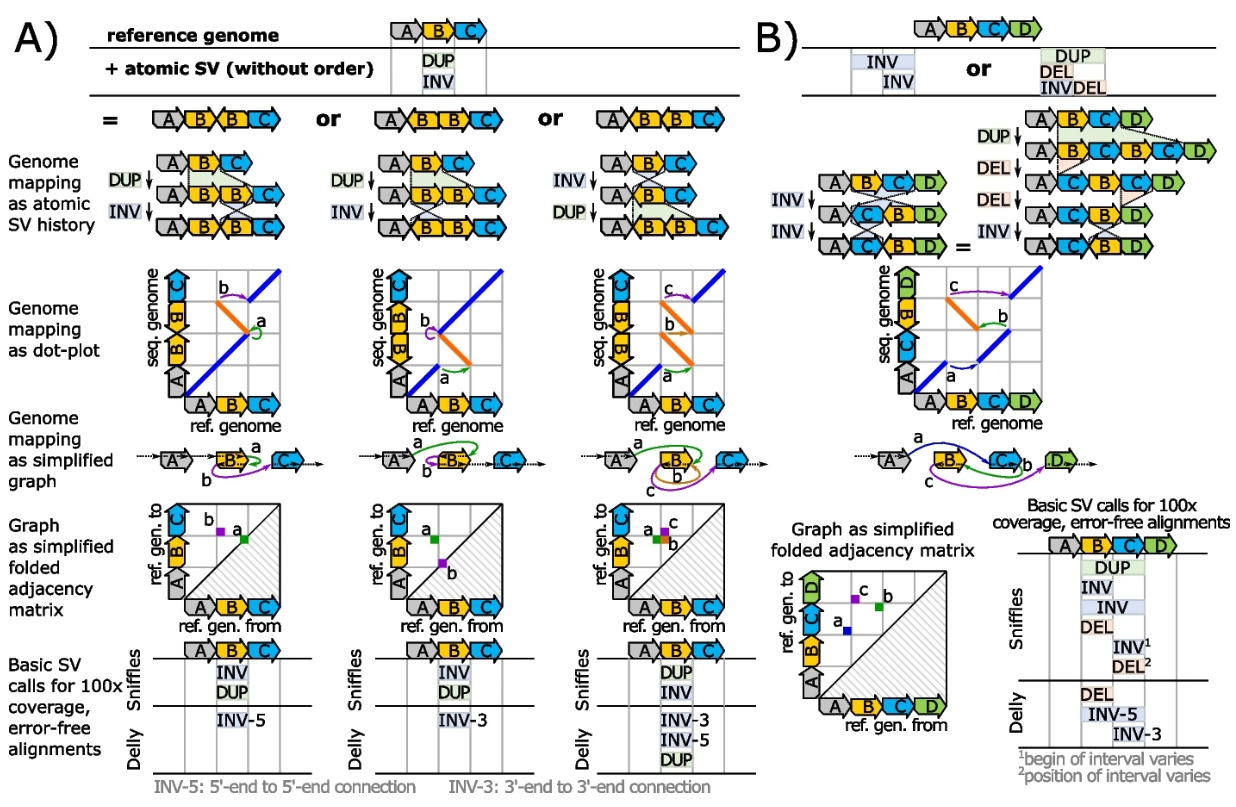
MSV:直接从reads发现结构变异
- 结构变异(SV)调用属于现代生物信息学的标准工具,用于识别和描述基因组的改变。
- 我们的工作提出了几种复杂的基因组重排,揭示了通过基本 SV 表示所固有的概念模糊性。我们从理论上和实践上将这些歧义联系起来,并提出一种基于图的方法来解决它们。
- 对于各种酵母基因组,我们实际上计算了图模型的邻接矩阵,并证明它们根据一个基因组提供了另一个基因组的高度准确的描述。
- 我们的方法的开源原型实现可在 MIT 许可证下获得,网址为 https://github.com/ITBE-Lab/MA 。
多响应孟德尔随机化检测复杂结果-暴露关系
Multi-response Mendelian randomization: Identification of shared and distinct exposures for multimorbidity and multiple related disease outcomes. Am J Hum Genet
英国伦敦帝国理工学院公共卫生学院流行病学和生物统计学系
是个不错的发现,有时间看看全文
- 孟德尔随机化 (MR) 的现有框架推断了一种或多种暴露对单一结果的因果影响。它并不是为了联合建模多种结果而设计的,因为有必要检测多种结果的原因,并且与模型多发病或其他相关疾病结果相关。
- 在这里,我们引入多响应孟德尔随机化 (MR2),这是一种专门为多种结果设计的 MR 方法,用于识别导致多个结果的暴露,或者相反,对不同响应产生影响的暴露。 MR2 使用稀疏贝叶斯高斯 copula 回归框架来检测因果效应,同时估计摘要级别结果之间的残差相关性,即无法由暴露解释的相关性,反之亦然。
- 我们从理论上和综合模拟研究中展示了不可测量的共享多效性如何在不考虑样本重叠的情况下导致结果之间的剩余相关性。我们还揭示了影响多种结果的非遗传因素如何影响它们的相关性。我们证明,通过考虑残差相关性,MR2 具有更高的能力来检测导致多个结果的共享暴露。与忽略相关响应之间的依赖性的现有方法相比,它还提供了更准确的因果效应估计。
- 最后,我们说明了 MR2 如何在考虑心脏代谢和脂质组学暴露的两个应用中检测五种心血管疾病的共同和不同因果暴露,并揭示反映心血管疾病之间已知关系的汇总级结果之间的残余相关性。
LSMMD-MA:扩展多模式数据集成以进行单细胞基因组学数据分析
LSMMD-MA: scaling multimodal data integration for single-cell genomics data analysis. Bioinformatics
Google Research的Brain Team
- 单细胞组学数据分析中的模态匹配,即在使用不同类型的基因组分析收集的数据集中匹配细胞已成为一个重要的问题,因为统一不同技术的观点有望产生生物学和临床发现。然而,单细胞数据集大小现在可以达到数十万到数百万个细胞,这对于大多数多模态计算方法来说仍然是遥不可及的。
- 我们提出了 LSMMD-MA,这是用于多模态数据集成的 MMD-MA 方法的大规模 Python 实现。在 LSMMD-MA 中,我们使用线性代数重新表述 MMD-MA 优化问题,并使用 KeOps(Python 中用于符号矩阵计算的 CUDA 框架)解决该问题。
- 我们表明,LSMMD-MA 在每种模式下可扩展至一百万个单元,比现有实现大两个数量级。
- LSMMD-MA 可在 https://github.com/google-research/large_scale_mmdma 免费获取,并存档在 https://doi.org/10.5281/zenodo.8076311。
- Bensz/ChatGPT:MMD-MA(Multimodal Data Integration using Maximum Mean Discrepancy and Mutual Attention)是一种用于多模态数据集成的方法。它结合了最大均值差异度量(Maximum Mean Discrepancy,MMD)和互相注意力机制(Mutual Attention)来实现多模态数据的融合。在多模态数据集成中,我们通常会面临不同模态之间的异构性、信息缺失以及噪声等挑战。MMD-MA方法旨在解决这些问题,并提供一个有效的方式来融合多个模态的信息。MMD是一种用于衡量两个概率分布之间差异的度量方法。它基于核函数将样本从不同分布映射到特征空间中,并计算两个分布在特征空间中的均值之差。MMD可以作为一个距离度量,用于比较不同模态数据的相似性。MMD-MA方法通过引入互相注意力机制来增强多模态数据的融合效果。互相注意力机制是一种利用注意力权重来加权聚合不同模态数据的方法。它能够自动学习每个模态数据对于融合结果的贡献程度,并根据这些权重进行数据融合。具体而言,MMD-MA方法包括以下步骤:(1)对每个模态数据应用特征提取器,将其转换为低维特征表示。(2)使用MMD方法计算不同模态数据之间的差异度量,并得到模态注意力权重。(3)使用注意力权重对每个模态数据进行加权,并利用加权后的特征进行融合。(4)将融合后的特征输入到分类器或回归器中进行目标预测。MMD-MA方法通过结合MMD和互相注意力机制,能够有效地融合多模态数据,并提供更全面、准确的信息,从而改善任务的性能。它在多模态场景下具有广泛的应用潜力,例如图像与文本的关联分析、音频与视频的情感识别等。
(~ ̄▽ ̄)~ 基于高通量数据的临床决策深度迁移学习:基准结果的综合调查
Deep transfer learning for clinical decision-making based on high-throughput data: comprehensive survey with benchmark results. Brief Bioinform. full pdf
了解一下文章的内容。与GSClassifier有关。
- 基于组学的数据的快速增长彻底改变了生物医学研究和精准医学,使机器学习模型能够开发出尖端性能。然而,尽管有大量可用的高通量数据,但这些模型的性能因缺乏足够的训练数据而受到阻碍,特别是在临床研究(体内实验)中。因此,将这些知识转化为临床实践,例如预测药物反应,仍然是一项具有挑战性的任务。迁移学习是一种很有前途的工具,它通过将知识从源域迁移到目标域来弥合数据域之间的差距。研究人员提出了利用临床前数据(小鼠、斑马鱼)的迁移学习来预测临床结果,突显了其巨大的潜力。
- 在这项工作中,我们对健康信息学和临床决策的深度迁移学习方法进行了全面的文献综述,重点关注高通量分子数据。之前的评论主要涵盖基于图像的迁移学习工作,而我们对迁移学习论文进行了更详细的分析。此外,我们根据交叉验证、数据分割和模型架构的不同评估设置来评估原始研究。
- 结果表明,这些迁移学习方法具有巨大的潜力;高通量测序数据和最先进的深度学习模型可以带来重要的见解和结论。此外,我们还通过统计和可视化探索了迁移学习论文中的各种数据集。
主动调控元件之间的超长程相互作用
Ultra-long-range interactions between active regulatory elements. Genome Res
- 增强子-启动子接触被认为与它们激活转录的能力有关。因此,研究促成这种染色质相互作用的因素对于理解基因调控非常重要。
- 在这里,我们从染色体构象捕获数据集中确定了数百万对顺式调控元件之间的接触频率,并分析了数百个 DNA 结合因子的集合,以在富集接触区域进行结合。该分析揭示了与活性转录相关的许多因素结合的位点上丰富的接触。
- 我们表明,独立于粘连蛋白和多梳的活性调控元件在脊椎动物和无脊椎动物基因组中在数十兆碱基的距离内相互作用,并且这种相互作用与活性相关并随活性而变化。然而,这些超长程相互作用并不依赖于 RNA 聚合酶 II 转录或单个转录辅助因子。通过模拟,我们表明染色质和多价结合因子的模型可以通过桥接诱导的聚类产生远程相互作用。
- 我们提出,顺式调控元件之间的长程相互作用至少由三个不同的过程驱动——粘连蛋白介导的环挤出、多梳接触和活性区域的聚集。
综述类
真核RNA解旋酶与人类疾病
- RNA 解旋酶是高度保守的蛋白质,使用三磷酸核苷来结合或重塑 RNA、RNA-蛋白质复合物或两者。 RNA解旋酶分为DEAD-box、DEAH/RHA、Ski2-like、Upf1-like和RIG-I家族,是真核RNA代谢中最大的一类活性酶——基因表达及其调控的几乎所有方面都涉及RNA解旋酶。这些酶的突变和失调与多种疾病有关,包括癌症和神经系统疾病。
- 在这篇综述中,我们讨论了 RNA 解旋酶的调控和功能机制及其在真核 RNA 代谢中的作用,包括转录调控、前 mRNA 剪接、核糖体组装、翻译和 RNA 衰变。我们重点介绍了将解旋酶结构、功能机制(例如局部链解旋、易位、绞盘、RNA 夹紧和置换 RNA 结合蛋白)和生物学作用联系起来的有趣模型,包括 RNA 解旋酶和通过液-液相分离形成的细胞凝聚物之间的新兴联系。
- 我们还讨论了 RNA 解旋酶与人类疾病的关联,以及最近为设计这些真核基因表达关键调节因子的小分子抑制剂所做的努力。
Apache Spark 分布式计算系统使用建议
Ten quick tips for bioinformatics analyses using an Apache Spark distributed computing environment. PLoS Comput Biol
- 一些科学研究涉及大量的生物信息学数据,这些数据无法在研究人员通常用于日常活动的个人计算机上进行分析,而是需要能够以分布式方式工作的有效计算基础设施。为此,分布式计算系统已成为分析大量生物信息学数据并在虚拟环境中生成相关结果的有用工具,其中软件可以执行数小时甚至数天,而不会影响研究人员的个人计算机或笔记本电脑。
- 即使分布式计算资源已成为多个生物信息学实验室的关键,但研究人员和学生常常以错误的方式使用它们,所犯的错误可能导致分布式计算机表现不佳,甚至可能产生错误的结果。
- 在此背景下,我们在此提出使用 Apache Spark 分布式计算系统进行生物信息学分析的十个快速提示:十个简单的指南,如果加以考虑,可以帮助用户避免常见错误,并帮助他们顺利地进行生物信息学分析。即使我们为初学者和学生设计了建议,专家也应该遵循它们。
- 我们认为我们的快速提示可以帮助任何人更有效地使用 Apache Spark 分布式计算系统,并最终帮助生成更好、更可靠的科学结果。
DDR、核酸与抗肿瘤免疫的互补性
The complementarity of DDR, nucleic acids and anti-tumour immunity. Nature
- 免疫检查点阻断(ICB)免疫疗法是特定癌症的一线治疗方法,但其疗效机制仍不完全清楚。此外,只有少数癌症患者能从 ICB 中受益,并且缺乏信息丰富的治疗反应生物标志物。得益于对 DNA 损伤反应 (DDR) 的深入了解,选择性地利用 DNA 损伤修复缺陷也是癌症的标准治疗方法。
- DDR和ICB紧密相连——有缺陷的DDR会产生免疫原性癌症新抗原,可以提高ICB治疗的疗效,而肿瘤突变负荷是对ICB反应的良好但不完善的生物标志物。 ICB 疗效的 DDR 研究最初侧重于对新抗原负担的影响。
- 然而,越来越多的证据表明,外源性 DNA 损伤或内源性过程(如 DNA 复制)产生的核酸的免疫原性效应使 ICB 功效变得复杂。化疗、放疗或选择性 DDR 抑制剂(如 PARP 抑制剂)可产生异常核酸,从而独立于新抗原诱导肿瘤免疫原性。独立于其免疫功能,免疫疗法的靶标(例如环 GMP-AMP 合酶 (cGAS) 或 PD-L1)可以与 DDR 或 DNA 修复机制相互作用,从而影响对 DNA 损伤剂的反应。
- 在这里,我们回顾了 DDR、核酸免疫原性和免疫治疗功效之间快速发展的多方面界面,重点关注 ICB。了解这些相互关联的过程可以解释 ICB 治疗的失败,并揭示癌症中新的可利用的治疗漏洞。最后,我们解决了尚未解答的主要问题和新的研究方向。
医学LLM
Large language models in medicine. Nat Med. full pdf
- 大语言模型 (LLM) 可以响应自由文本查询,而无需接受相关任务的专门培训,这引起了人们对其在医疗保健环境中使用的兴奋和担忧。 ChatGPT 是一种生成式人工智能 (AI) 聊天机器人,是通过LLM的复杂微调而产生的,其他工具也通过类似的开发过程不断涌现。
- 在这里,我们概述了 ChatGPT 等 LLM 应用程序的开发方式,并讨论了如何在临床环境中利用它们。我们考虑LLM的优势和局限性及其提高医学临床、教育和研究工作效率和效果的潜力。 LLM 聊天机器人已经部署在一系列生物医学领域,取得了令人印象深刻但好坏参半的结果。
- 这篇综述可以作为感兴趣的临床医生的入门读物,他们将确定LLM技术是否以及如何在医疗保健中使用,以造福患者和从业者。
流行病学类
不良童年经历与终身健康的关联
- 随着可持续发展目标的出现,全球儿童健康领域的重点已从降低死亡率转向改善健康、营养和发展成果(通常以人力资本来衡量)。人们对发育生物学和神经科学的了解不断加深,凸显了不良环境暴露(统称为不良童年经历(ACE))对健康结果的重要性。 ACE 与健康和发育的短期、中期和长期负面后果相关,其影响可能是倍增的,特别是在敏感性和发育可塑性的关键时期。其中一些影响因气候变化、冲突和人口流离失所等新出现的全球威胁而加剧。
- 在本次综述中,我们讨论了将 ACE 与健康结果联系起来的关键机制,并考虑了预防和减轻其影响的有前景的策略,重点介绍了低收入和中等收入国家项目的证据。最后,我们强调需要尽早承认 ACE 并提供涵盖卫生、教育、妇女赋权和社会保护等关键部门的一揽子干预措施。
洛杉矶地区多元化群体的疾病风险和医疗保健利用
Disease risk and healthcare utilization among ancestrally diverse groups in the Los Angeles region. Nat Med
- 由于共同的遗传和环境因素,个体的疾病风险受到其所属人群的影响。临床护理中小规模人群的研究对于识别和减少健康差异以及制定个性化干预措施非常重要。
- 为了评估小规模人群的临床诊断和医疗保健利用模式,我们利用了 35,968 名患者的遗传数据和电子病历,作为加州大学洛杉矶分校阿特拉斯社区健康计划的一部分。我们使用血统身份来定义个体集群,血统身份是一种遗传相关性形式,利用由于共同祖先而产生的共享基因组片段。
- 我们总共发现了 376 个聚集性病例,其中包括加勒比非裔、波多黎各人、黎巴嫩基督徒、伊朗犹太人和古吉拉特裔患者的聚集性病例。我们的分析发现了疾病诊断和聚类之间的 1,218 个显着关联以及与专科就诊之间的 124 个显着关联。我们还检查了致病等位基因的分布,发现 189 个重要等位基因在特定簇中频率较高,其中包括许多不定期纳入人群筛查工作的等位基因。
- 总体而言,这项工作增进了对受研究社区健康的理解,并可以为进一步研究健康不平等问题奠定基础。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍
