本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- 免疫细胞类型之间的相互作用促进免疫特征的进化
- 人类病理学的多模态生成式人工智能助手
- 重编程结合人转铁蛋白受体的 AAV 衣壳介导全脑基…
- 人类媒介微生物相互作用图谱揭示了致病机制
- 体内编辑肺干细胞以实现小鼠持久基因校正
前言
本文是前沿快讯的第52期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。从第18期开始,“肿瘤转移类”、“肿瘤代谢类”等将不再更新,而是合并至其它分类。原有的流行病学类也改为科普类。
本期有以下知识点值得关注:
CNS类
免疫细胞类型之间的相互作用促进免疫特征的进化
Interactions between immune cell types facilitate the evolution of immune traits | Nature. Nature
- 自然选择进化的一个重要先决条件是影响适应度的个体特征的变异。系统产生可选择变异的能力,称为进化性,因此极大地影响进化速率。免疫系统属于哺乳动物中进化最快的组成部分,但免疫特征变异的来源在很大程度上仍然未知。
- 在这里,我们表明,免疫系统进化性的一个重要决定因素是其组织成由不同免疫细胞类型代表的相互作用的模块。通过分析来自 Collaborative Cross6 的 54 种遗传多样性小鼠品系的骨髓中的免疫细胞变异,我们发现免疫细胞频率的变异是多基因的,并且许多相关基因通过增殖、迁移和细胞死亡等细胞内在功能参与稳态平衡。
- 然而,我们还发现了与特定细胞类型的频率相关的基因,这些基因在不同的细胞类型中表达,在我们所说的细胞反式中发挥作用。脊椎动物的进化记录表明,与细胞反式相关的基因面临较弱的阴性选择,从而增加了免疫系统的稳健性和进化性。这种现象在人类血液中也同样可以观察到。
- 我们的研究结果表明,免疫系统不同组成部分之间的相互作用提供了一个表型空间,突变可以在其中产生变异而不会造成太大损害,这强调了模块化在复杂系统进化中的作用。
人类病理学的多模态生成式人工智能助手
A Multimodal Generative AI Copilot for Human Pathology | Nature. Nature
波士顿哈佛医学院布莱根妇女医院病理科Faisal Mahmood团队
- 计算病理学领域在任务特定预测模型和任务无关的自监督视觉编码器的开发方面取得了显着进展。然而,尽管生成人工智能 (AI) 呈爆炸式增长,但针对病理学构建通用、多模式 AI 助手的研究仍然有限。
- 在这里,我们推出 PathChat,一种用于人类病理学的视觉语言多面手人工智能助手。我们通过采用病理学基础视觉编码器,将其与预训练的大型语言模型相结合,并根据超过 456,000 种不同的视觉语言指令(包括 999,202 个问答轮)对整个系统进行微调来构建 PathChat。我们将 PathChat 与几种多模态视觉语言 AI 助手和 GPT4V 进行比较,GPT4V 为商用多模态通用 AI 助手 ChatGPT-4 提供支持。
- PathChat 在来自不同组织来源和疾病模型的病例的多项选择诊断问题上取得了最先进的性能。此外,通过使用开放式问题和人类专家评估,我们发现整体 PathChat 对与病理学相关的各种查询产生了更准确且更适合病理学家的响应。
- 作为一种交互式通用视觉语言 AI Copilot,可以灵活处理视觉和自然语言输入,PathChat 可以在病理学教育、研究和人机交互临床决策中找到有影响力的应用。
重编程结合人转铁蛋白受体的 AAV 衣壳介导全脑基因传递
- 开发能够有效地将基因传递到整个人类中枢神经系统(CNS)的载体将扩大可治疗遗传疾病的范围。我们设计了一种腺相关病毒 (AAV) 衣壳 BI-hTFR1,它可以结合人转铁蛋白受体 (TfR1)(一种在血脑屏障上表达的蛋白质)。
- BI-hTFR1 主动转运穿过人脑内皮细胞,并且相对于 AAV9,在人 TFRC 敲入小鼠的 CNS 中提供了 40 至 50 倍的报告基因表达。增强的趋向性是中枢神经系统特异性的,并且在野生型小鼠中不存在。当用于递送 GBA1(GBA1 的突变会导致戈谢病并与帕金森病相关)时,与 AAV9 相比,BI-hTFR1 显着增加了大脑和脑脊液葡萄糖脑苷脂酶活性。
- 这些发现确立了 BI-hTFR1 作为人类中枢神经系统基因治疗的潜在载体。
人类媒介微生物相互作用图谱揭示了致病机制
An atlas of human vector-borne microbe interactions reveals pathogenicity mechanisms – PubMed. Cell
- 媒介传播疾病是全世界死亡的主要原因,并造成大量未得到满足的医疗需求。与宿主细胞外蛋白(“外蛋白组”)结合的病原体代表了媒介传播疾病病因学中的一个重要界面。
- 在这里,我们使用细菌选择来阐明高通量宿主微生物相互作用 (BASEHIT)——一种能够询问微生物与 3,324 种人类外蛋白相互作用的技术——来分析 82 个人类病原体样本的相互作用组,其中包括 30 种节肢动物传播的病原体菌株以及 8 株相关的非媒介传播病原体。由此产生的图谱揭示了 1,303 个假定的相互作用,包括数百个在发病机制中具有潜在作用的配对,包括细胞入侵、组织定植、免疫逃避和宿主感知。随后的功能研究发现莱姆病螺旋体将表皮生长因子识别为转录调节的环境线索,并且细胞内病原体和硫氧还蛋白之间的保守相互作用促进细胞侵袭。
- 总之,该相互作用组图谱提供了对微生物发病机制的分子水平见解,并揭示了下一代治疗的潜在宿主导向靶标。
体内编辑肺干细胞以实现小鼠持久基因校正
In vivo editing of lung stem cells for durable gene correction in mice | Science. Science
- 体内基因组校正有望产生持久的疾病治疗方法;然而,有效的干细胞编辑仍然具有挑战性。
- 在这项工作中,我们证明优化的肺靶向脂质纳米粒子(LNP)能够在干细胞中进行高水平的基因组编辑,从而产生持久的反应。在可激活的 tdTomato 小鼠中静脉注射基因编辑 LNP,实现了 >70% 的肺干细胞编辑,在 >80% 的肺上皮细胞中维持 tdTomato 表达达 660 天。解决囊性纤维化 (CF),NG-ABE8e 信使 RNA (mRNA)–sgR553X LNP 介导 >95% 囊性纤维化跨膜电导调节因子 (CFTR) DNA 校正,恢复原发性患者来源支气管上皮细胞中的 CFTR 功能,相当于 F508del 的 Trikafta,纠正了 CF 小鼠 50% 肺干细胞中的肠道类器官和 R553X 无义突变。
- 这些发现引入了支持 LNP 的组织干细胞编辑,用于疾病修饰基因组校正。
噬菌体尾样细菌素抑制病原菌集合种群中的竞争者
- 了解影响植物病原体传播的因素对于制定疾病预防策略至关重要。细菌病原体必须克服植物免疫力,与微生物竞争,并抵抗噬菌体在植物中定殖。宿主遗传学影响病原体抑制,但周围微生物群的作用仍不清楚。噬菌体(噬菌体)、细菌病毒和噬菌体衍生元件是选择性杀死细菌的广泛存在的实体。这些元素从噬菌体祖先中重新利用,以共存的细菌菌株为目标,有可能塑造微生物群落。尽管它们普遍存在,但它们对植物微生物群和病原体传播的影响仍然很大程度上未知。
- 我们之前的工作表明,与农业和临床病原体爆发不同,拟南芥野生种群是由没有优势菌株的遗传多样性假单胞菌种群定植的。即使在单个植物中,绿色假单胞菌的感染也包括多种共存菌株。是什么阻止单一致病谱系的传播?宿主免疫多样性可能有助于病原体多样性,但植物微生物组也可能发挥作用。鉴于噬菌体和噬菌体衍生元件的普遍存在及其在假单胞菌种群中的菌株特异性杀伤活性,我们假设对噬菌体成分的敏感性差异可以抑制特定菌株。
- 我们发现了丰富的病毒簇,在致病菌株中是保守的。该簇不编码完整的噬菌体,而是编码尾素,这是细菌用来杀死细菌竞争对手的噬菌体衍生元素。每个致病性假单胞菌菌株都携带几种不同的尾素变体之一,其目标是共存病原体外膜中的可变多糖。对历史植物标本的分析表明,相同的尾素和外膜变体在假单胞菌种群中持续存在了至少两个世纪,这表明一组确定的尾素单倍型和受体的持续使用。我们的结果支持一个模型,其中共存假单胞菌菌株的尾素和外膜在野生拟南芥种群中协同进化。不同的tailocin和外膜单倍型在时间和空间上一直保持着。
- 集合群体中存在有限的尾素单倍型集可能反映了有限的耐药机制。我们的研究结果为识别尾素对不同菌株的特异性以及确定这种特异性机制的可能性提供了路线图。 Tailocin 疗法类似于噬菌体疗法,有望成为传统抗生素的替代品。初步研究证明其在各种植物和动物模型中抑制病原体的功效。然而,与任何抗菌治疗一样,可能会出现耐药性。我们的结果表明,利用病原体遗传多样性的“tailocin 鸡尾酒”可以通过同时针对集合种群来减轻耐药性。
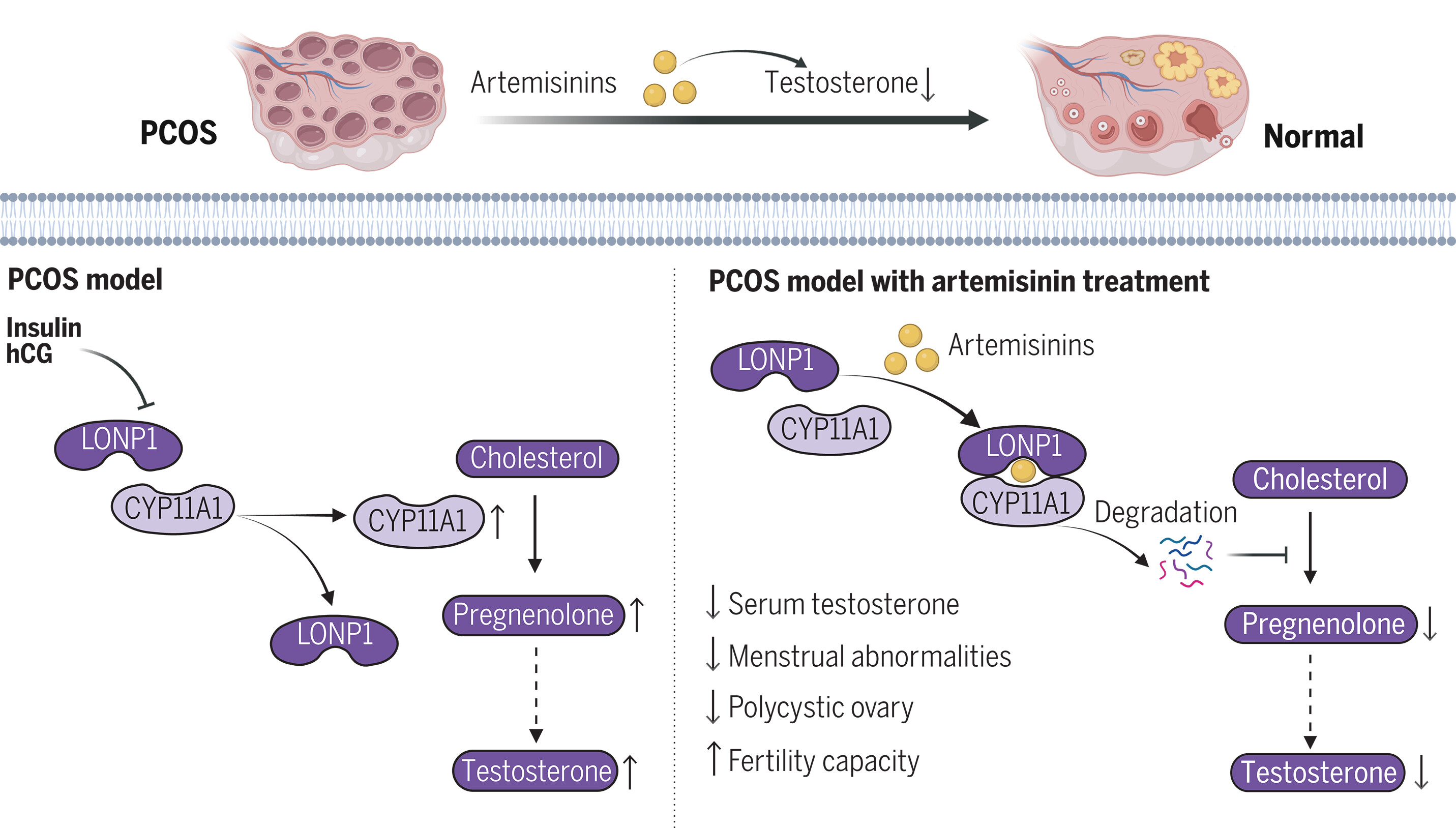
青蒿素通过介导 LONP1-CYP11A1 相互作用改善多囊卵巢综合征
Artemisinins ameliorate polycystic ovarian syndrome by mediating LONP1-CYP11A1 interaction | Science. Science
- 多囊卵巢综合征 (PCOS) 是一种常见的生殖内分泌疾病,影响育龄妇女的 10% 至 13%,其特征是高雄激素血症、排卵功能障碍、多囊卵巢形态,并且常常伴有相关的代谢紊乱。雄激素过多是导致多囊卵巢综合征表型特征的关键因素。尽管多囊卵巢综合症的患病率很高,但对这种复杂综合征的药物干预却遇到了巨大的挑战。目前可用于多囊卵巢综合症的治疗选择有限,并且主要针对特定症状的管理。因此,迫切需要开发创新的治疗策略。
- 青蒿素源自青蒿植物,因其抗疟功效而被广泛认可。我们之前已经证明,青蒿素及其衍生物具有通过激活产热脂肪细胞来增强能量消耗和胰岛素敏感性的能力,从而防止饮食引起的肥胖和代谢紊乱。在这项研究中,我们通过评估青蒿素衍生物对睾酮水平、动情周期和多囊卵巢形态的影响,探讨了青蒿素在啮齿类 PCOS 样模型和人类 PCOS 患者中的治疗潜力。使用体外和体内方法,我们研究了青蒿素对卵巢睾酮合成的影响。确定青蒿素的直接靶点,以阐明青蒿素调节睾酮合成的机制。
- 我们发现,青蒿素类似物蒿甲醚在 PCOS 样啮齿动物模型中表现出对高雄激素血症、不规则动情周期、多囊卵巢形态和低生育力的显着改善。青蒿素通过抑制卵巢睾酮合成来抑制高雄激素血症。相对定量蛋白质组学分析显示,细胞色素 P450 家族 11 亚家族 A 成员 1 (CYP11A1)(催化雄激素合成第一步的酶)是受青蒿素影响最显着减少的蛋白质。进一步研究表明,青蒿素诱导CYP11A1降解,导致卵巢雄激素合成受到抑制。在没有 CYP11A1 的情况下,这种抑制作用会减弱。
- 从机制上讲,青蒿素直接靶向lon肽酶1(LONP1),增强LONP1和CYP11A1之间的相互作用,促进LONP1催化CYP11A1的降解。相反,雄激素诱导剂破坏 LONP1 和 CYP11A1 之间的结合;此外,LONP1 在 PCOS 中下调,导致 CYP11A1 水平升高和雄激素合成增加。蛋白质对接模拟和随后的功能实验表明,青蒿素对 CYP11A1 水平的抑制作用很大程度上取决于其与 LONP1 蛋白水解结构域的直接结合。与青蒿素的功能一致,LONP1 过度表达强烈抑制卵巢中雄激素的产生。最后,进行了一项试点临床试验,以证实青蒿素对 PCOS 患者的治疗效果。我们发现双氢青蒿素治疗可有效改善高雄激素血症,降低抗苗勒氏管激素水平,改善多囊卵巢形态,并有助于 PCOS 患者月经正常化。
- 我们的数据证明了青蒿素在缓解啮齿类动物模型和人类患者中与多囊卵巢综合症相关的症状方面的功效。青蒿素直接与 LONP1 结合,启动 LONP1 与 CYP11A1 之间的相互作用,进而促进 CYP11A1 的降解,从而抑制卵巢雄激素合成,抑制 PCOS。相反,雄激素诱导剂会破坏 LONP1-CYP11A1 相互作用并加重 PCOS。总的来说,我们的研究结果凸显了青蒿素作为综合治疗多囊卵巢综合征的有效药物的巨大潜力。这一发现阐明了 LONP1 和 CYP11A1 之间以前未知的相互作用,青蒿素可以增强这种相互作用来控制雄激素合成,从而为通过靶向 LONP1-CYP11A1 相互作用来干预 PCOS 开辟了途径。
代谢不灵活促进肝脏再生过程中的线粒体健康
Metabolic inflexibility promotes mitochondrial health during liver regeneration | Science. Science
- 线粒体电子传递链 (ETC) 功能障碍常见于获得性人类疾病,包括代谢相关肝病。在肝脏再生过程中,增殖的肝细胞相互竞争,使适应性增强的细胞更容易为再生器官的组成做出贡献。尽管最近的研究表明线粒体ETC功能可以影响干细胞行为,但尚不清楚ETC是否会影响肝损伤后的肝细胞增殖,从而有助于器官状况和功能的恢复。
- 我们使用小鼠线粒体代谢物分析和同位素示踪技术来研究肝细胞在稳态和再生条件下的代谢反应。然后,我们使用一组针对线粒体 ETC 的遗传小鼠模型来剖析每个单独的 ETC 复合物(I 至 V)对肝再生的贡献。通过这种方法,我们旨在检查野生型(WT)和ETC功能障碍肝细胞在再生过程中的相对适应性,并确定增殖肝细胞中线粒体健康的调节机制。
- 我们发现小鼠肝细胞在肝脏再生过程中需要功能性 ETC 来增殖并与 WT 肝细胞竞争。在没有 ETC 的情况下,小鼠肝脏迅速积累脂肪酸,导致脂肪变性。我们还观察到,在 ETC 突变肝脏再生过程中,胆管细胞向肝细胞的转分化受到刺激。代谢追踪研究表明,WT 肝脏依靠外周脂肪储存的动员和氧化来维持增殖过程中乙酰辅酶 A 的水平。在 ETC 突变的肝脏中,脂肪酸氧化受到抑制,导致脂肪积累和乙酰辅酶 A 的产生减少。值得注意的是,线粒体复合物 I 不是肝细胞增殖所必需的,这表明复合物 I 不是肝细胞再生中 ETC 的主要电子供体。
- 当脂肪在 ETC 功能障碍的情况下积聚时,由于 PDK4(丙酮酸氧化的负调节因子)的诱导表达和 ACSS2 表达的减少,非脂肪酸来源(例如丙酮酸或乙酸盐)中乙酰辅酶 A 的生成受到抑制。 这种代谢不灵活性(无法切换到替代营养素来生成乙酰辅酶A)迫使人们依赖脂肪酸氧化,从而选择具有功能性 ETC 的增殖肝细胞。为了测试这个模型,我们抑制或删除 PDK4 以重新启用丙酮酸氧化为乙酰辅酶 A。在缺乏 PDK4 活性的情况下,ETC 功能障碍的肝细胞能够在肝脏再生过程中增殖。
- 我们的结果支持一个模型,即调节肝脏营养利用的网络拓扑编码代谢不灵活性,从而在组织再生过程中促进线粒体健康。具体来说,在 ETC 功能障碍的情况下脂肪酸的积累会抑制替代底物生成乙酰辅酶 A。我们将脂肪积累下游的 PDK4 表达确定为控制增殖肝细胞代谢不灵活性的关键调控事件。尽管代谢灵活性在很大程度上被认为有益于生物体的生存和功能,但我们的模型表明,小鼠肝脏可以利用代谢不灵活性来促进增殖细胞群的整体健康。
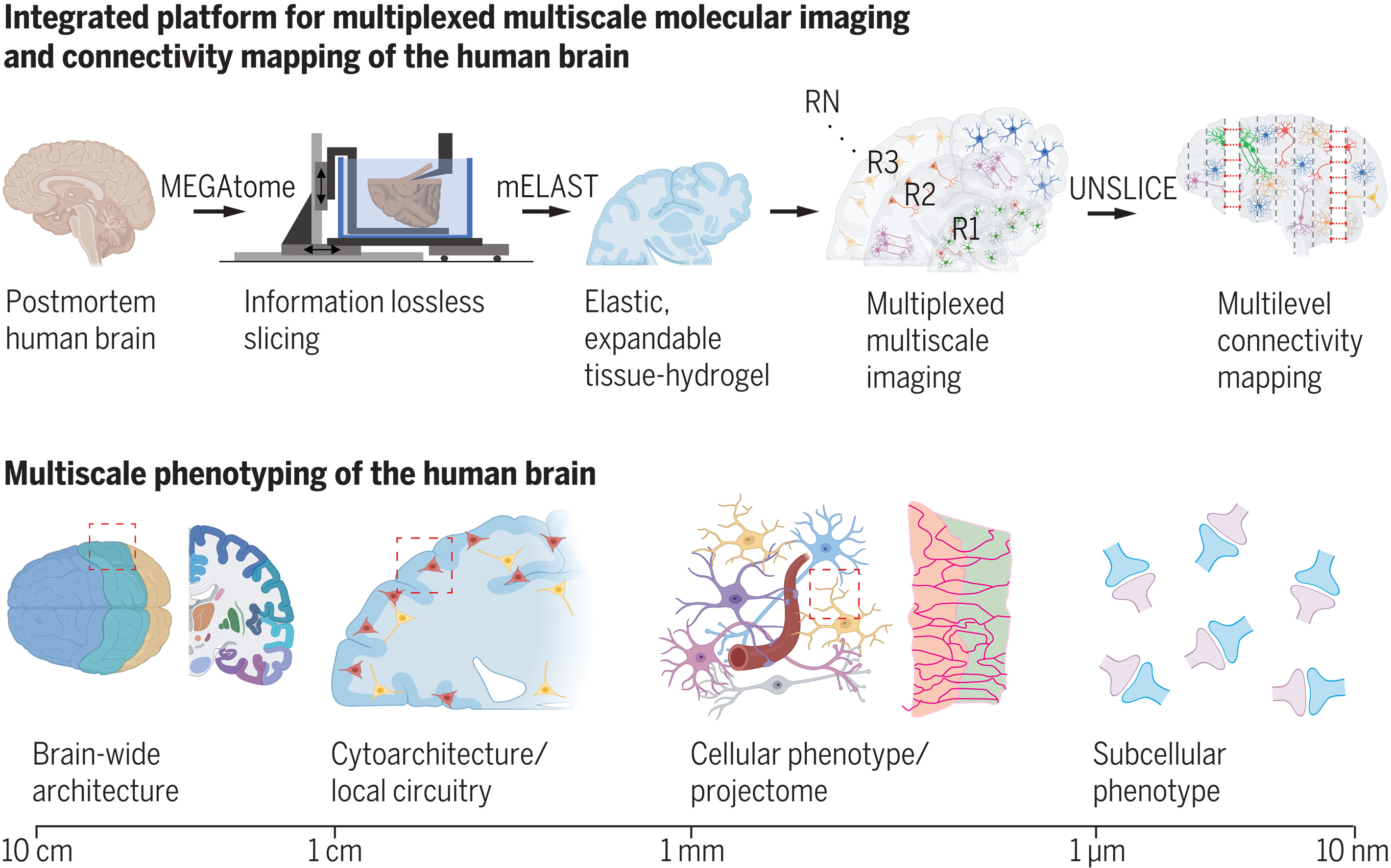
人脑多尺度分子成像和表型分析集成平台
Integrated platform for multiscale molecular imaging and phenotyping of the human brain | Science. Science
麻省理工学院 (MIT) 医学工程与科学研究所
- 了解人体器官功能和功能障碍需要详细绘制细胞的解剖和分子结构及其全器官连接性。成像和分子分析技术的进步极大地丰富了我们对人体器官内功能区域和细胞的解剖组织及其分子特性的理解。然而,我们仍然缺乏能够以整体方式捕获单个细胞的多尺度多组学特性及其全器官连接性的技术。
- 我们开发了一个可扩展的技术平台,用于同时绘制从同一组织获取的细胞的器官范围结构和高维特征,包括分子、形态和连接信息。该平台由一个能够保持连接的组织切片的机械装置、一种用于工程组织理化特性以实现多重多尺度分子成像的化学技术,以及一个用于单细胞投影组图谱的计算工具组成。我们完全集成了机械、化学和计算工具,以实现人体器官规模组织的高度多重多尺度分子表型分析。
- MEGAtome(机械增强型大尺寸无磨损振动切片机)能够对超大型生物系统进行精确切片,同时最大限度地减少连接信息的丢失,这要归功于优化刀片振动控制的多自由度 (DOF) 系统。 MEGAtome 切片和光片成像促进了超大规模样本的高通量分子图谱绘制,例如完整的冠状人脑板和队列规模的动物器官阵列。基于水凝胶的集成组织处理方法称为 mELAST(可放大纠缠链增强可拉伸组织水凝胶),将生物组织转化为弹性、透明和可膨胀的水凝胶,同时保留内源性生物分子和纳米级细胞结构。与 SWITCH(化学物质相互作用时间和动力学的系统范围控制)介导的快速染色方法相结合,mELAST 能够对完整的人脑组织进行高度多重的多尺度成像。 UNSLICE(通过互连的切割纤维端点的连接来统一相邻的切片组织)促进了准确的板间配准,以使用免疫标记的细胞类型特异性纤维作为地标在单纤维水平上重建切片组织块。随着我们增加数据集的维度,我们策略的迭代性质使得连通性映射的准确性得以持续提高。我们应用集成技术平台在多个尺度上分析人类阿尔茨海默病(AD)病理学,揭示了不同的病理特征,包括细胞类型分布、形态特征、神经元纤维取向和化学突触分布的差异。利用 UNSLICE,我们展示了人脑中单纤维分辨率的可扩展神经投影映射,揭示了表达病理蛋白的神经纤维的投影模式。
- 我们的技术平台能够以前所未有的分辨率和速度对人脑规模组织中的细胞进行可扩展且完全集成的结构和分子表型分析。我们设想该平台将能够对大量人类和动物大脑进行整体分析,从而促进我们对物种间同源性、群体差异和疾病特异性特征的理解。此外,我们的方法能够绘制单神经元投影组及其与分子表达谱的整合。这一独特的特征将使我们能够阐明神经回路的组织原理及其在人脑中的疾病特异性改变,从而增进我们对疾病机制的理解。
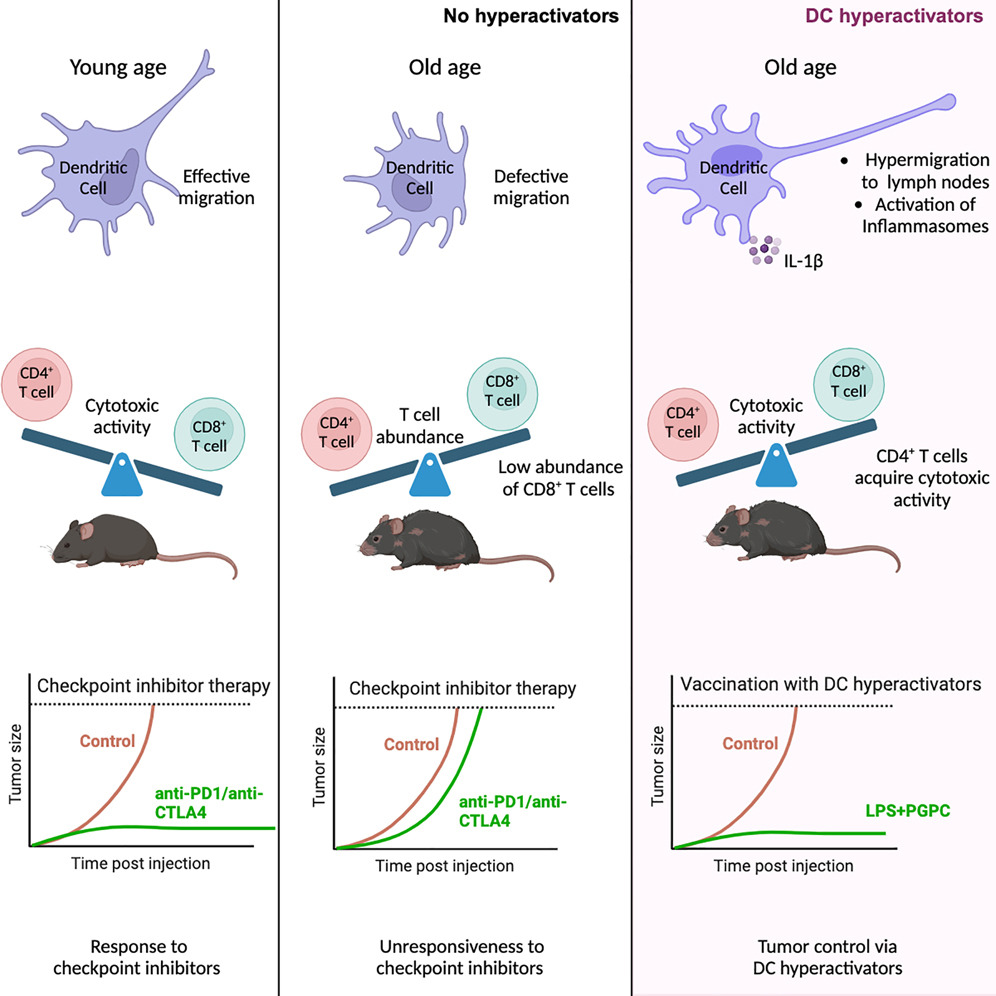
纠正树突状细胞中与年龄相关的缺陷使 CD4+ T 细胞能够根除肿瘤
哈佛医学院和波士顿儿童医院胃肠病学部
- 晚年宿主防御缺陷与免疫细胞活性的变化有关,这表明免疫治疗方法需要考虑年龄特异性。
- 在这项研究中,我们发现基于PD-1和CTLA4的癌症免疫疗法无法根除老年小鼠的肿瘤。这种抗肿瘤活性缺陷与两种已知的与年龄相关的免疫缺陷相关:全身幼稚 CD8+ T 细胞丰度减少和树突状细胞 (DC) 迁移活性弱。
- 我们发现了一种称为 DC 过度激活剂的疫苗佐剂,它可以纠正老年人的 DC 迁移缺陷。含有肿瘤抗原和 DC 过度激活剂的疫苗可诱导具有细胞溶解活性的 1 型辅助 T (TH1) CD4+ T 细胞,从而驱动老年小鼠的抗肿瘤免疫。当在生命早期施用时,DC 过度激活剂是唯一确定的能够引发持续到老年的抗肿瘤 CD4+ T 细胞的佐剂。
- 这些结果提出了通过 DC 操作纠正与年龄相关的免疫缺陷的可能性。
时间分辨命运图谱将肠道上隐窝区识别为 Lgr5+ 隐窝基底柱状细胞的起源
- 在流行的模型中,Lgr5+细胞是唯一能够通过难以捉摸的上隐窝转运放大(TA)中间体向上迁移后代来维持稳态上皮再生的肠道干细胞(ISC)。
- 在这里,我们在假定的 TA 细胞位置鉴定了一个由 Fgfbp1 标记的增殖性上隐窝群体,其转录与 Lgr5+ 细胞不同。
- 使用动力学报告器进行时间分辨命运图谱和 Fgfbp1-CreERT2 谱系追踪,我们确定 Fgfbp1+ 细胞是多能的,并产生 Lgr5+ 细胞,与其 ISC 功能一致。 Lgr5+ 细胞耗竭后,Fgfbp1+ 细胞还可维持上皮再生。我们证明,由上隐窝细胞产生的 FGFBP1 是隐窝增殖和上皮稳态的重要因素。
- 我们的研究结果支持这样一种模型:组织再生起源于上隐窝 Fgfbp1+ 细胞,这些细胞产生沿隐窝-绒毛轴双向繁殖的后代,并作为隐窝基部 Lgr5+ 细胞的来源。
MTFP1控制线粒体融合以调节内膜质量控制并维持mtDNA水平
- 线粒体动力学在细胞命运决定以及控制 mtDNA 水平和分布中发挥着关键作用。然而,将线粒体膜重塑和质量控制与 mtDNA 拷贝数 (CN) 调节联系起来的分子机制仍然难以捉摸。
- 在这里,我们证明线粒体内膜 (IMM) 蛋白线粒体裂变过程 1 (MTFP1) 负向调节 IMM 融合。此外,通过调节 MTFP1 水平来操纵线粒体融合会导致 mtDNA CN 调节。
- 从机制上讲,我们发现 MTFP1 抑制线粒体融合,以将受损的 IMM 子域与网络的其余部分隔离并排除。随后,外周裂变确保它们分离成富含 MTFP1 的小线粒体 (SMEM),这些线粒体以自噬依赖性方式进行降解。
- 值得注意的是,依赖于 MTFP1 的 IMM 质量控制对于基础类核回收至关重要,因此对于维持细胞内足够的 mtDNA 水平至关重要。
单细胞新生RNA测序揭示了协调的全局转录
Single-cell nascent RNA sequencing unveils coordinated global transcription | Nature. Nature. full html
美国麻省理工学院生物学系、科赫综合癌症研究所Phillip A. Sharp团队
- 转录是基因表达的主要调控步骤。启动子和增强子的不同转录起始产生来自基因的稳定 RNA 和来自增强子的不稳定 RNA。新生 RNA 捕获和测序测定可同时测量细胞群中的基因和增强子活性。然而,关于转录的时间调控和增强子-基因协调的基本问题仍未得到解答,这主要是因为缺乏对主动转录的单细胞视角。
- 在这项研究中,我们提出了 scGRO–seq——一种使用点击化学的新型单细胞新生 RNA 测序分析——并揭示了整个基因组的协调转录。
- 我们证明了转录的情景性质和功能相关基因的共转录。 scGRO-seq 可以通过直接量化单个细胞中转录 RNA 聚合酶来估计突发大小和频率,并可以利用复制依赖性非多腺苷酸化组蛋白基因转录来阐明细胞周期动态。 scGRO–seq 的单核苷酸空间和时间分辨率能够识别增强子和基因网络。我们的结果表明,超级增强子的转录爆发先于相关基因的爆发。
- 通过深入了解全局转录的动态性质以及转录信号的起源和传播,我们证明了 scGRO-seq 研究转录调控机制以及增强子在基因表达中的作用的能力。
饮食诱导和精子携带的线粒体 RNA 的表观遗传
Epigenetic inheritance of diet-induced and sperm-borne mitochondrial RNAs | Nature. Nature
和之前的父系肠道菌群影响子代性状的发现有类似之处;但该影响的比重有多大?
- 精子拥有一个复杂且对环境敏感的小非编码 RNA (sncRNA) 池,它影响后代发育和成年表型。附睾中的精子是否直接容易受到环境因素的影响尚不完全清楚。
- 在这里,我们使用两种不同的孕前急性高脂肪饮食范例来剖析附睾与睾丸对精子 sncRNA 库和后代健康的贡献。我们发现附睾精子(但不包括发育中的生殖细胞)对环境敏感,并将线粒体 tRNA (mt-tRNA) 及其片段 (mt-tsRNA) 识别为精子传播因子。在人类中,精子中的 mt-tsRNA 与体重指数相关,而父亲在受孕时超重会使后代肥胖风险加倍,并损害代谢健康。
- 对涉及线粒体功能的基因突变小鼠的精子 sncRNA 测序及其野生型后代的代谢表型分析表明,mt-tsRNA 的上调是线粒体功能障碍的下游。遗传杂交两细胞胚胎的单胚胎转录组学证明了受精时 mt-tRNA 的精子到卵母细胞的转移,并表明它们参与了早期胚胎转录的控制。
- 我们的研究支持受孕时父亲健康对后代新陈代谢的重要性,表明mt-tRNA是饮食诱导的和精子传播的,并证明在生理环境中,受精时精子线粒体RNA从父亲到后代的转移。
用于能量转换的大面积自修复嵌段共聚物膜
Large-area, self-healing block copolymer membranes for energy conversion | Nature. Nature
- 膜广泛用于海水淡化、电池和透析等应用中的分离过程,在我们的经济和社会的关键部门中至关重要1。大多数技术开发的膜都是基于固体聚合物并充当被动屏障,其传输特性由其化学成分和纳米结构决定。尽管此类膜无处不在,但事实证明,独立地最大化选择性和渗透性具有挑战性,从而导致这些相关特性之间的权衡。自组装生物膜,其中屏障和运输功能是解耦的 3,4,为解决这个问题提供了灵感 5,6。
- 在这里,我们介绍了一种自组装策略,该策略使用水性两相系统的界面来模板化和稳定分子薄(约 35nm)仿生嵌段共聚物双层,其可扩展面积可以超过 10cm2 而没有缺陷。这些膜具有自我修复能力,其对离子通过的屏障功能(比电阻约为1 MΩ cm2)接近于磷脂膜。这些膜的流动性使得分子载体能够直接进行功能化,使钾离子沿着浓度梯度输送,并且对钠离子具有精细的选择性。
- 这种离子选择性使得能够在模拟电射线电器官的装置中从 NaCl 和 KCl 的等摩尔溶液产生电力。
下降网络将命令信号转化为群体运动控制
Descending networks transform command signals into population motor control | Nature. Nature
挺特别的研究发现
- 为了将意图转化为行动,运动指令必须通过下行神经元(DN)从大脑传递到下游运动回路。其中包括足以驱动行为的小组类似命令的神经元——其电路机制仍不清楚。
- 在这里,我们展示了果蝇中类似命令的 DN 直接招募额外 DN 的网络来协调需要主动控制许多身体部位的行为。具体来说,我们发现,以前认为类似命令的 DN 会单独驱动行为;事实上它们共同激活了更大的 DN 群体。
- 连接组分析和实验操作表明,这种功能性募集可以通过大脑中命令型 DN 和互连 DN 网络之间的直接兴奋性连接来解释。下降种群招募对于行为控制是必要的:具有许多下游下降伙伴的 DN 需要网络共同激活来驱动完整的行为,并且在它们不存在的情况下仅驱动简单的刻板运动。这些 DN 网络驻留在特定行为的集群中,彼此相互抑制。
- 这些结果支持了一种类似命令的下降控制机制,其中行为是通过招募越来越大的 DN 网络来生成的,这些 DN 网络通过组合多个运动子程序来构成行为。
衰老的神经胶质细胞与线粒体功能障碍和脂质积累有关
Senescent glia link mitochondrial dysfunction and lipid accumulation | Nature. Nature
- 衰老是一种与许多哺乳动物物种的衰老和年龄发病相关的细胞状态。衰老细胞迅速促进伤口愈合并防止肿瘤形成;但它们也具有促炎作用,因此会长期加剧组织衰退。尽管衰老细胞是抗衰老治疗的积极靶标,但这些细胞为何在体内形成、它们如何影响组织衰老以及消除它们的效果仍不清楚。
- 在这里,我们识别了衰老果蝇大脑中自然发生的衰老神经胶质细胞,并破译了它们的起源和影响。使用激活蛋白 1 (AP1) 活性来筛选衰老,我们确定衰老的神经胶质细胞可能因神经元线粒体功能障碍而出现。反过来,衰老的神经胶质细胞促进非衰老神经胶质细胞的脂质积累;在培养的衰老人类成纤维细胞中也观察到类似的效果。靶向衰老神经胶质细胞中的 AP1 活性可减轻衰老生物标志物,延长果蝇寿命和健康寿命,并防止脂质积累。然而,这些好处是以大脑氧化损伤增加为代价的,而且神经元线粒体功能仍然很差。
- 总而言之,我们的结果绘制了体内自然发生的衰老神经胶质细胞的轨迹,并表明这些细胞与关键的衰老现象有关:线粒体功能障碍和脂质积累。
POT1 在人类端粒处招募并调节 CST-Polα/引物酶
POT1 recruits and regulates CST-Polα/primase at human telomeres – PubMed. Cell
- 端粒的维持需要通过端粒酶延伸富含G的端粒重复链,并通过Polα/引物酶填充合成富含C的端粒重复链。在端粒处,Polα/引物酶与单链 DNA 结合复合物 Ctc1/Stn1/Ten1 (CST) 结合。与端粒酶突变一样,影响 CST-Polα/引物酶的突变会导致病理性端粒缩短并导致端粒生物学紊乱,Coats plus (CP)。
- 我们确定了与庇护蛋白异二聚体 POT1/TPP1 结合的人 CST 的低温电子显微镜结构,揭示了 CST 如何被 POT1 募集到端粒上。
- 我们的研究结果表明,POT1 铰链磷酸化是 CST 募集所必需的,并且该复合物是通过涉及 CP 中突变的几个残基的保守相互作用形成的。我们的结构和生化数据表明,磷酸化的 POT1 使 CST-Polα/引物酶保持在非活性、自抑制状态,直到端粒酶延长端粒末端。
- 我们建议 POT1 的去磷酸化将 CST-Polα/引物酶释放到活性状态,通过填充合成完成端粒复制。
DNMT3A突变驱动的克隆造血促进炎性骨质流失
Clonal hematopoiesis driven by mutated DNMT3A promotes inflammatory bone loss – PubMed. Cell
- 不确定潜能克隆造血 (CHIP) 是由造血祖细胞中与衰老相关的获得性突变引起的,这些突变表现出克隆扩张并产生表型改变的白细胞。
- 我们将 CHIP-DNMT3A 突变与 4,946 名社区居住成年人中较高的牙周炎和牙龈炎症患病率联系起来。为了模拟 DNMT3A 驱动的 CHIP,我们使用了具有杂合功能丧失突变 R878H(相当于人类热点突变 R882H)的小鼠。
- Dnmt3aR878H/+骨髓(BM)细胞的部分移植导致突变细胞克隆扩增至骨髓和淋巴谱系,并且BM中破骨细胞前体和周围破骨细胞巨噬细胞的丰度升高。受体小鼠中 DNMT3A 驱动的克隆造血促进了自然发生的牙周炎,并加剧了实验诱导的牙周炎和关节炎,这与破骨细胞生成增强、IL-17 依赖性炎症和中性粒细胞反应以及调节性 T 细胞免疫抑制活性受损有关。雷帕霉素治疗抑制了 DNMT3A 驱动的克隆造血和随后的牙周炎。
- DNMT3A 驱动的 CHIP 代表了一种可治疗的适应不良造血促进炎症性骨质流失的状态。
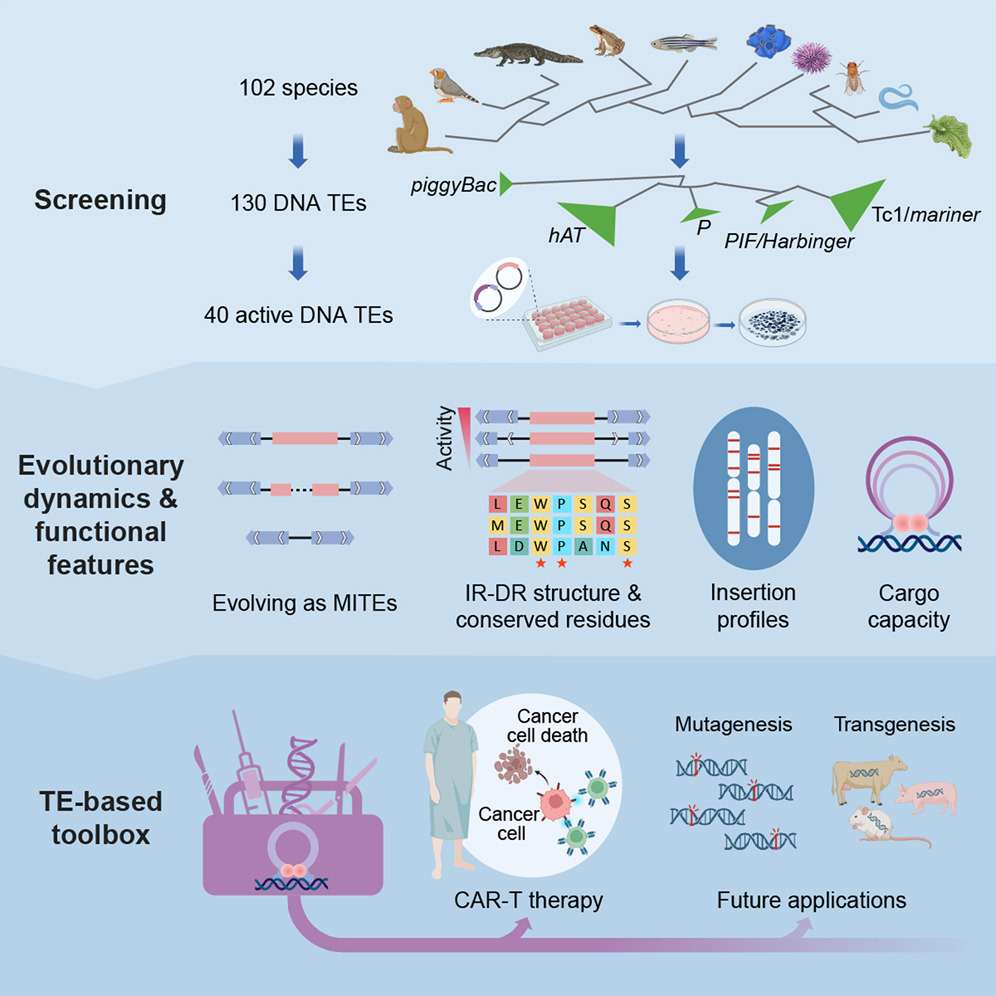
对人类细胞中 130 个 DNA 转座子的异源调查突显了它们的功能差异并扩展了基因组工程工具箱
- 实验研究中的DNA转座元件(DNA transposable elements, TEs)规模有限,导致对影响转座活动、进化动态和作为基因组工程工具的应用潜力的理解不足。
- 我们从102个后生动物基因组中预测出130个活跃的DNA TEs,并在人类细胞中评估了它们的活性。我们鉴定出40个活跃的(具有整合能力的)TEs,超过了此前发现的TEs的累计数量(20个)。
- 通过这一统一的比较数据,我们发现Tc1/mariner超家族展示出较高的活性,这可能解释了它们普遍的水平转移。进一步的TEs功能表征揭示了如插入偏好等特征的额外分化。值得注意的是,在用于血液和实体瘤的CAR-T治疗中,我们鉴定出的最活跃的DNA TE Mariner2_AG(MAG),大大超过了两种广泛使用的载体,即慢病毒载体和基于TE的载体SB100X。
- 总体而言,这项研究突出了DNA TEs的多样化转座特征和进化动态,并增加了TE工具箱的多样性。
通过剪接体抑制捕获人类细胞的全能性
Capturing totipotency in human cells through spliceosomal repression – PubMed. Cell
- 合子裂解生成全能性卵裂球。在人类的8细胞卵裂球中,合子基因组激活(zygotic genome activation, ZGA)发生,以启动个体发育程序。然而,捕捉和维持人类细胞的全能性面临重大挑战。
- 在此,我们实现了培养人类全能性卵裂球样细胞(human totipotent blastomere-like cells, hTBLCs)。我们发现剪接抑制可以暂时将人类多能干细胞重新编程为ZGA样细胞(ZGA-like cells, ZLCs),这些细胞在长期传代后转变为稳定的hTBLCs。
- 与已报道的8细胞样细胞(8-cell-like cells, 8CLCs)不同,ZLCs和hTBLCs广泛沉默多能基因。有趣的是,ZLCs激活了一组特定的ZGA特异性基因,而hTBLCs富含前ZGA特异性基因。在自发分化过程中,hTBLCs重新进入中间的ZLC阶段,并进一步生成上胚层(epiblast, EPI)、原始内胚层(primitive endoderm, PrE)和滋养外胚层(trophectoderm, TE)样谱系,有效地重现了人类着床前发育。hTBLCs具有胚胎和非胚胎发育的潜力,可以在体外自主生成类囊胚结构而无需外部细胞信号。
- 总之,我们的研究提供了人类细胞全能性的关键标准和见解。
核糖应激反应驱动紫外线介导的细胞死亡
The ribotoxic stress response drives UV-mediated cell death – PubMed. Cell
- 虽然紫外线 (UV) 辐射会损伤 DNA,引发 DNA 损伤反应 (DDR),但它也会损伤 RNA,引发转录组范围内的核糖体碰撞并引发核糖毒性应激反应 (RSR)。然而,这些途径在决定细胞命运方面的相对贡献、时间和调节尚不清楚。
- 在这里,我们使用时间分辨磷酸化蛋白质组学、化学遗传学、单细胞成像和生化方法来创建细胞响应紫外线损伤时激活的信号事件的时间顺序图谱。我们发现紫外线诱导的细胞凋亡是由 RSR 激酶 ZAK 介导的,而不是通过 DDR 介导的。
- 我们确定了调节 ZAK 介导的细胞凋亡的两个负反馈模块:(1)GCN2 激活限制核糖体碰撞并减弱 ZAK 介导的 RSR;(2)ZAK 活性导致磷酸化蛋白自磷酸化及其随后的降解。
- 这些事件将 ZAK 的活性调整至碰撞水平,以建立稳态、耐受性和死亡机制,揭示了其作为核酸损伤的细胞哨兵的关键作用。
通过机器学习在全微生物组中发现抗菌肽
Discovery of antimicrobial peptides in the global microbiome with machine learning – PubMed. Cell. full html
复旦大学类脑智能科学技术研究院、宾夕法尼亚大学-佩雷尔曼医学院-精神病学和微生物学系-医学和治疗学研究所、生物医学信息学研究所-机器生物学组
抗菌肽流程:Prodigal + Macrel + MMSeqs2
- 迫切需要新型抗生素来应对抗生素耐药性危机。我们提出了一种基于机器学习的方法来预测全球微生物组中的抗菌肽 (AMP),并利用来自环境和宿主相关栖息地的 63,410 个宏基因组和 87,920 个原核基因组的庞大数据集来创建 AMPSphere,这是一个包含 863,498 个非冗余肽,其中很少有与现有数据库匹配的肽。
- AMPSphere 提供了对肽的进化起源的见解,包括通过较长序列的复制或基因截断,我们观察到 AMP 的产生因栖息地而异。
- 为了验证我们的预测,我们在体外和体内合成并测试了 100 种 AMP,以对抗临床相关的耐药病原体和人类肠道共生体。共有 79 种肽具有活性,其中 63 种针对病原体。这些活性 AMP 通过破坏细菌膜而表现出抗菌活性。
- 总之,我们的方法鉴定了近一百万个原核 AMP 序列,这是抗生素发现的开放获取资源。
一年生鳉鱼轴的形成:节点和β-连环蛋白在没有Huluwa预模式的情况下调节形态发生
- 鱼类和两栖动物的轴形成通常始于母体基因产物的预模式。然而,每年的鳉鱼胚胎发生对预模式模型提出了挑战,因为卵裂球分散然后聚集形成胚层和体轴。
- 我们发现,huluwa(一种被认为通过稳定 β-连环蛋白来打破对称性的预模式因子)在 Nothobranchius furzeri 中被截短且失活。核β-连环蛋白不是选择性地稳定在囊胚的一侧,而是在形成聚集体的细胞中积累。阻断 β-连环蛋白活性或 Nodal 信号传导会破坏聚集体形成和胚层规范。节点信号传导协调细胞迁移,确立了该信号传导途径的早期作用。
- 这些结果揭示了与已建立的轴形成机制的惊人背离:Huluwa 介导的预模式是可有可无的,β-catenin 和 Nodal 调节形态发生。
人γ-分泌酶底物识别和裂解的分子机制
Molecular mechanism of substrate recognition and cleavage by human γ-secretase | Science. Science
- γ-分泌酶连续切割具有 99 个残基的淀粉样蛋白前体蛋白 C 末端片段 (APP-C99),产生不同长度的淀粉样蛋白-β (Aβ) 肽。大多数切割的步长为三个残基。
- 为了阐明其潜在机制,我们确定了分别与 APP-C99、Aβ49、Aβ46 和 Aβ43 结合的人 γ-分泌酶的原子结构。在所有情况下,底物都显示出相同的结构特征:跨膜 α-螺旋、三残基接头和与早老素 1 (PS1) 形成杂合 β-折叠的 β-链。蛋白水解裂解发生在底物 β 链之前。每次裂解后,底物 α 螺旋解旋并易位一圈,并形成新的 β 链。
- 该机制与现有的生化数据一致,并且可以解释γ-分泌酶对其他底物的裂解。
稀有密码子重新编码,用于哺乳动物细胞中有效的非规范氨基酸掺入
Rare codon recoding for efficient noncanonical amino acid incorporation in mammalian cells | Science. Science
听上去很不错
- 对非规范氨基酸 (ncAA) 进行基因编码的能力使蛋白质具有改进的或以前未知的特性。然而,哺乳动物细胞中现有的策略依赖于引入空白密码子来掺入 ncAA,这种方法效率低下并限制了其广泛应用。
- 在本研究中,我们开发了一种稀有密码子重新编码策略,利用 TCG 密码子的相对稀有性,通过系统工程和大数据模型预测实现高选择性和高效的 ncAA 掺入。
- 我们强调了该策略的广泛实用性,可将数十种 ncAA 掺入野生型蛋白表达水平的各种功能蛋白中,以及在哺乳动物细胞中合成具有多达 6 个位点 ncAA 或 4 个不同 ncAA 的蛋白质下游应用。
哺乳动物脂肪组织产热的两阶段进化
Two-stage evolution of mammalian adipose tissue thermogenesis | Science. Science
- 棕色脂肪组织 (BAT) 是一种发热器官,表达产热解偶联蛋白 1 (UCP1),以在冷应激期间维持较高的体温。 BAT 生热作用被认为是哺乳动物的一项重要特征,但其进化起源尚不清楚。
- 我们发现,有袋动物的脂肪组织在约 1.5 亿年前从真兽类哺乳动物中分化出来,表达一种非产热 UCP1 变体,该变体由部分转录组 BAT 特征控制,与真兽类米色脂肪组织中发现的相似。
- 我们发现,重建的真兽类祖先 UCP1 序列表现出典型的产热活性,而兽类祖先 UCP1 则不具有产热活性。因此,哺乳动物脂肪组织产热作用可能经历了两个不同的阶段,其中共同兽类祖先的产热前阶段将UCP1表达与脂肪组织和热应激联系起来。
- 我们认为,在第二阶段,UCP1 在真兽类中获得了其产热功能,因此哺乳动物 BAT 产热作用仅在与有袋动物分化后才发生。
细胞结构塑造幼稚 T 细胞反应
Cellular architecture shapes the naïve T cell response | Science. Science
比较清奇的角度 ~
- T 细胞消除受感染的细胞并提供长期保护,防止同一病原体的再次感染。其中的核心是通过 T 细胞受体 (TCR) 检测外来抗原。抗原结合后,表达相同 TCR 的幼稚 T 细胞克隆群会分化为具有细胞毒性能力的效应 T 细胞和记忆 T 细胞,从而提供长期免疫。这种多样化模式在群体水平上是可重复的,但单个幼稚 T 细胞在抗原反应期间会走上不同的轨迹。尽管已知初始 T 细胞的命运受到外部因素(包括抗原亲和力和可用性)的影响,但受控抗原环境中克隆相同的 T 细胞仍然表现出不同的反应和异质分化轨迹。这表明细胞内在的决定因素和细胞间变异在调节 T 细胞分化中具有潜在作用。细胞异质性的一个日益受到重视的来源是细胞形态的变化和亚细胞成分的空间组织(即细胞的结构)。因此,我们系统地研究了原代人类和小鼠 T 细胞的结构异质性,以及这种异质性在预先确定初始 T 细胞对抗原的反应强度和分化轨迹中的潜在作用。
- 通过将高通量荧光显微镜与基于深度学习的单细胞图像分析相结合,我们报告说,T 细胞 (TARCH) 细胞结构的异质性预先确定了它们响应抗原刺激的 TCR 信号强度和随后的分化轨迹。这种结构异质性由三类定义:形态极化 T 细胞 (TP) 或球形 T 细胞,不带(TO;“常规”)或带(TØ;“条纹”)深部核膜内陷 (NEI)。这些 NEI 在空间上集中了细胞机器,包括内质网、线粒体和核孔复合体。这三类 T 细胞的结构平衡随着 T 细胞的成熟、激活和分化而变化。
- 在分子定义的 T 细胞亚群中,其结构特征具有高度可重复性,在人类和小鼠体内,60% 的初始 CD8 T 细胞采用 TØ 结构。体内病毒感染后幼稚 CD8 T 细胞的分化与效应期 TØ 的减少和 TP 结构的增加有关,随后在感染后 28 天重建 TØ 高的病毒特异性记忆细胞群。此外,抗原 T 细胞激活后,TARCH 亚群表现出不同的分子和功能反应:TØ 结构在依赖于激活后最初几分钟内钙流入增加的机制中表现出更强的 TCR 信号传导。这与已知影响 TCR 信号强度的蛋白质的表达无关,并且可以通过药理学抑制钙池操纵的钙进入以及通过 TØ 向 TO 结构的转换来抑制。结构上解析的离体单细胞命运追踪显示,与 TO 细胞相比,TØ 细胞更早开始细胞分裂,并增殖形成更大的细胞集落,这些细胞具有下调的 TCF1 和上调的肿瘤坏死因子-α (TNF-α) 和干扰素-γ (IFN-γ) 细胞因子表达,与效应样 T 细胞表型一致。相比之下,具有 TO 结构的细胞在 TCR 刺激下分裂速度较慢,并产生 TCF1+ 细胞,这表明它们会优先分化为具有记忆表型的 T 细胞。
- 我们的数据表明,细胞结构是早期 TCR 信号传导以及随后个体幼稚 T 细胞响应抗原的细胞命运决定的预先决定因素。将细胞结构理解为 T 细胞生物学的表型维度,可能能够预测和优化 T 细胞反应,以防止感染和对抗疾病。
肿瘤免疫类
癌细胞中的核苷酸代谢促进 UDP 驱动的巨噬细胞串扰,促进免疫抑制和免疫治疗耐药性
- 许多癌症患者对免疫疗法有抵抗力。在这里,我们确定了编码嘧啶补救途径酶胞苷脱氨酶(CDA)的基因是几种免疫治疗耐药肿瘤中最上调的代谢基因之一。
- 我们发现癌细胞中的 CDA 有助于尿苷二磷酸 (UDP) 库。细胞外 UDP 通过其受体 P2Y6 劫持免疫抑制性肿瘤相关巨噬细胞 (TAM)。癌细胞中 CDA(或 TAM 中的 P2Y6)的药理或基因抑制会破坏 TAM 介导的免疫抑制,促进细胞毒性 T 细胞进入,并提高耐药性胰腺导管腺癌中抗程序性细胞死亡蛋白 1(抗 PD-1)治疗的敏感性。 PDAC)和黑色素瘤模型。相反,CDA 耗尽的 PDAC 或抗 PD-1 反应性结直肠肿瘤中 CDA 过表达或全身 UDP 给药(重新)建立耐药性。
- 在患有 PDAC 的个体中,癌细胞中高 CDA 水平与 TAM 增加、细胞毒性 T 细胞降低以及可能的抗 PD-1 耐药性相关。在泛癌单细胞图谱中,CDAhigh 癌细胞与 T 细胞细胞毒性功能障碍和 P2RY6high TAM 相匹配。
- 总体而言,我们建议 CDA 和 P2Y6 作为癌症免疫治疗的潜在靶点。
临床类
人工智能和放射科医生在 MRI 前列腺癌检测 (PI-CAI) 中的应用:一项国际配对、非劣效性验证性研究
- 人工智能(AI)系统可以通过减轻日益增加的工作量、防止过度诊断和减少对经验丰富的放射科医生的依赖来帮助前列腺癌的诊断途径。我们的目的是研究人工智能系统在 MRI 检测临床上显着的前列腺癌方面的性能,并与使用前列腺成像报告和数据系统 2.1 版 (PI-RADS 2.1) 的放射科医生以及大规模多学科常规实践中的护理标准进行比较。
- 在这项国际配对、非劣效性验证性研究中,我们训练并外部验证了一个人工智能系统(在国际联盟内开发),用于使用来自 9129 名患者的 10207 例 MRI 检查的回顾性队列来检测格里森 2 级或以上癌症患者。在这些检查中,来自荷兰三个中心(11个地点)的9207个案例用于训练和调整,来自荷兰和挪威四个中心(12个地点)的1000个案例用于测试。与此同时,我们利用 PI-RADS (2.1) 对 62 名放射科医生(20 个国家的 45 个中心;阅读前列腺 MRI 的经验中位数为 7 [IQR 5-10] 年)进行了一项多读者、多病例观察者研究,对 400 项配对 MRI 检查进行了研究测试队列。主要终点是与使用 PI-RADS (2.1) 的所有读取器相比以及与历史放射学读数相比,AI 系统的灵敏度、特异性和受试者工作特征曲线下面积 (AUROC)。多学科常规实践(即借助患者病史和同行咨询的护理标准)。使用组织病理学和至少 3 年(中位 5 [IQR 4-6] 年)的随访来建立参考标准。统计分析计划预先设定了非劣效性的主要假设(考虑 0·05 的裕度)和在非劣效性得到确认的情况下对人工智能系统的优越性的次要假设。该研究已在 ClinicalTrials.gov 注册,NCT05489341。
- 在2012年1月1日至2021年12月31日期间进行的10207例检查中,2440例经组织学证实患有格里森2级或更大的前列腺癌。在将 AI 系统与参与读者研究的放射科医生进行比较的 400 个测试案例子集中,AI 系统显示出统计上优越且非劣势的 AUROC 为 0·91 (95% CI 0·87-0·94 ; p<0·0001),与 AUROC 为 0·86 (0·83-0·89) 的 62 名放射科医生池相比,两侧 95% Wald CI 的差异下限AUROC 为 0·02。在所有读者的平均 PI-RADS 3 或更高操作点上,AI 系统在相同特异性下检测到的格里森 2 级或以上癌症病例增加了 6·8%(57·7%,95% CI 51·6-) 63·3),或在相同敏感性下,假阳性结果减少 50·4%,格里森 1 级癌症病例减少 20·0% (89·4%,95% CI 85·3-92·9)。在将人工智能系统与多学科实践中的放射学读数进行比较的所有 1000 个测试案例中,未确认非劣效性,因为人工智能系统表现出较低的特异性(68·9% [95% CI 65·3-72·4] ] 与 69·0% [65·5-72·5]) 在与 PI-RADS 3 或更高操作点相同的灵敏度 (96·1%、94·0-98·2) 下。特异性差异的两侧 95% Wald CI 的下限 (-0·04) 大于非劣效性界限 (-0·05),并且达到低于显着性阈值的 p 值 (p<0 ·001)。
- 平均而言,在检测有临床意义的前列腺癌方面,AI 系统优于使用 PI-RADS (2.1) 的放射科医生,并且与护理标准相当。这样的系统显示出成为初级诊断环境中的支持工具的潜力,为患者和放射科医生带来了一些相关的好处。需要前瞻性验证来测试该系统的临床适用性。
新辅助纳武单抗或纳武单抗加 LAG-3 抑制剂 relatlimab 治疗可切除食管/胃食管交界癌:Ib 期试验和 ctDNA 分析
- 胃食管癌的动态和免疫检查点抑制剂(ICI)临床反应的驱动因素仍然知之甚少。双重程序性细胞死亡蛋白 1 (PD-1) 和淋巴细胞激活基因 3 (LAG-3) 抑制的潜在协同活性可能有助于改善这些肿瘤的免疫治疗反应。
- 我们报告了一项 Ib 期试验,该试验在 32 名可切除的 II 期/III 期胃食管癌患者中评估了新辅助纳武单抗(A 组,n = 16)或纳武单抗-relatlimab(B 组,n = 16)联合放化疗的效果。 -深入评估病理、分子和功能性免疫反应。主要终点是安全性;次要终点是可行性;探索性终点包括病理完全缓解(pCR)和主要病理缓解(MPR)、无复发生存期(RFS)和总生存期(OS)。
- 该研究在 A 组中达到了主要安全终点,但 B 组需要进行修改以减轻毒性。 A 组的 pCR 和 MPR 率为 40% 和 53.5%,B 组为 21.4% 和 57.1%。最常见的不良事件是疲劳、恶心、血小板减少和皮炎。总体而言,2 年 RFS 和 OS 率分别为 72.5% 和 82.6%。较高的基线程序性细胞死亡配体 1 (PD-L1) 和 LAG-3 表达与更深的病理反应相关。
- 对循环肿瘤 DNA (ctDNA) 的探索性分析表明,ICI 诱导后 ctDNA 检测不到的患者,术前和术后的 RFS 和 OS 显着延长; ctDNA 清除率反映了新抗原特异性 T 细胞反应。
- 我们的研究结果提供了对联合 PD-1 和 LAG-3 阻断治疗胃食管癌的安全性的见解,并强调了 ctDNA 分析在新辅助 ICI 期间动态评估全身肿瘤负荷的潜力,这可能为未来的干预打开治疗窗口。 ClinicalTrials.gov 注册号:NCT03044613。
联合抗 PD-1、HDAC 抑制剂和抗 VEGF 治疗 MSS/pMMR 结直肠癌:一项随机 2 期试验
中肿徐瑞华
- 染色质的表观遗传修饰,包括组蛋白乙酰化和肿瘤血管生成,在创建免疫抑制肿瘤微环境中发挥着关键作用。
- 在随机 2 期 CAPability-01 试验中,我们研究了程序性细胞死亡蛋白 1 (PD-1) 单克隆抗体信迪利单抗与组蛋白脱乙酰酶抑制剂 (HDACi) 西达本胺联合/不联合单克隆抗体贝伐珠单抗治疗不可切除的化疗难治性局部晚期或转移性微卫星稳定/熟练错配修复(MSS/pMMR)结直肠癌患者。 48 名患者被随机分配至双联组(信迪利单抗和西达本胺,n = 23)或三联组(信迪利单抗、西达本胺和贝伐单抗,n = 25)。
- 整个研究人群的 18 周无进展生存率 (PFS)(18wPFS 率)的主要终点率为 43.8%(48 人中的 21 人)。次要终点结果包括中位 PFS 为 3.7 个月、总体缓解率为 29.2%(48 例中的 14 例)、疾病控制率为 56.3%(48 例中的 27 例)以及中位缓解持续时间为 12.0 个月。中位总生存时间的次要终点尚未成熟。
- 与双联组相比,三联组的结果显着改善,18wPFS 率更高(64.0% 对比 21.7%,P = 0.003),总体缓解率更高(44.0% 对比 13.0%,P = 0.027),中位 PFS 率更长(7.3 个月与 1.5 个月,P = 0.006)。
- 在三联组和双联组中观察到的最常见的治疗出现的不良事件包括蛋白尿、血小板减少症、中性粒细胞减少症、贫血、白细胞减少症和腹泻。有两例与治疗相关的死亡(肝衰竭和肺炎)。
- 对患者肿瘤样本的bulk RNA 测序数据的分析表明,三联体组合增强了 CD8+ T 细胞浸润,从而产生了免疫活性更强的肿瘤微环境。
- 我们的研究表明,PD-1 抗体、HDACi 和 VEGF 抗体的组合可能是 MSS/pMMR 晚期结直肠癌患者的一种有前景的治疗方案。 ClinicalTrials.gov 注册:NCT04724239。
Botensilimab 加 balstilimab 治疗复发/难治性微卫星稳定转移性结直肠癌:1 期试验
- 微卫星稳定转移性结直肠癌(MSS mCRC;错配修复熟练)此前对免疫检查点阻断反应不佳。 Botensilimab (BOT) 是一种 Fc 增强型多功能 anti-CTLA-4 抗体,旨在扩大对冷/免疫原性差的实体瘤(例如 MSS mCRC)的治疗。
- 一项正在进行的扩展 1 期研究正在评估含或不含巴斯蒂利单抗(BAL;抗 PD-1 抗体)的 BOT。主要终点是安全性和耐受性,在研究的剂量递增部分和 MSS mCRC 患者中分别进行了评估(使用联合剂量递增/剂量扩展数据)。次要终点包括研究者评估的 RECIST 1.1 版确认的客观缓解率 (ORR)、疾病控制率 (DCR)、缓解持续时间 (DOR) 和无进展生存期 (PFS)。
- 在这里,我们展示了 148 名接受过多次治疗的 MSS mCRC 患者(其中 6 名来自剂量递增队列;142 名来自剂量扩展队列)接受 BOT 和 BAL 治疗的结果,其中 101 名患者被认为可通过至少 6 个月的治疗来评估疗效跟进。 89% 的 MSS mCRC 患者发生治疗相关不良事件 (TRAE) (131/148),最常见的是疲劳 (35%, 52/148)、腹泻 (32%, 47/148) 和发热 (24%, 36/148),没有报告 5 级 TRAE,并且由于 TRAE 导致的停药率为 12%(18/148;数据完全成熟)。在可评估疗效的人群 (n = 101) 中,ORR 为 17% (17/101;95% 置信区间 (CI),10-26%),DCR 为 61% (62/101;95% CI,51 -71%)。中位随访时间为 10.3 个月(范围:0.5-42.6),未达到中位 DOR(NR;95% CI,5.7 个月-NR),中位 PFS 为 3.5 个月(95% CI,2.7-4.1 个月)月;数据持续成熟)。
- BOT 与 BAL 的组合表现出可控的安全性,没有新的免疫介导的安全信号,并通过持久的反应鼓励临床活动。 ClinicalTrials.gov 标识符:NCT03860272。
共享新抗原疫苗联合免疫检查点阻断治疗晚期转移性实体瘤:1 期试验中期结果
- 引发针对肿瘤特异性新抗原的细胞毒性 T 细胞反应的治疗性疫苗有望为癌症患者提供长期临床益处。
- 在这里,我们评估了一种治疗性疫苗的安全性和耐受性,该疫苗编码了 20 种共同的新抗原,这些新抗原源自选定的常见致癌驱动突变,作为正在进行的针对晚期/转移性实体瘤患者的 1/2 期研究的主要终点。次要终点包括免疫原性、总体缓解率、无进展生存期和总体生存期。如果患者的肿瘤表达疫苗中包含的与人类白细胞抗原匹配的肿瘤突变之一,则选择符合条件的患者,其中大多数患者(18/19)携带 KRAS 突变。
- 该疫苗方案由黑猩猩腺病毒 (ChAd68) 和自扩增 mRNA (samRNA) 以及免疫检查点抑制剂伊匹单抗 (ipilimumab) 和纳武单抗 (nivolumab) 组成,显示出良好的耐受性,观察到的与治疗相关的不良事件与预期的急性炎症一致基于病毒载体的疫苗和免疫检查点封锁,大多数为 1/2 级。两名患者经历了 3/4 级严重的治疗相关不良事件,也是剂量限制性毒性。
- 总体缓解率为 0%,中位无进展生存期和总生存期分别为 1.9 个月和 7.9 个月。相对于患者肿瘤表达的 KRAS 新抗原,T 细胞反应偏向于疫苗中编码的人类白细胞抗原匹配的 TP53 新抗原,这表明以前未知的新抗原免疫优势等级可能会影响多表位共享新抗原疫苗的治疗效果。
- 这些数据促使开发出一种专门针对 KRAS 衍生新抗原的优化疫苗,该疫苗正在临床研究第 2 阶段的一部分患者中进行评估。 ClinicalTrials.gov 注册号:NCT03953235。
高危儿科癌症的精准引导治疗
Precision-guided treatment in high-risk pediatric cancers – PubMed. Nat Med
- 最近的研究表明,精准医学可以为儿童癌症患者找到新的治疗策略。然而,尚不清楚哪些患者将从精准引导治疗(PGT)中受益最多。
- 在此,我们报告了 384 名高危儿科癌症患者(预期治愈率低于 30%)的连续数据,这些患者对零儿童癌症精准医学计划 PREcISion Medicine for Children with Cancer 进行了至少 18 个月的随访(棱镜)试验。
- 共有 256 名患者 (67%) 接受了 PGT 建议,110 名患者 (29%) 接受了推荐治疗。与标准护理(26% 对比 12%;P = 0.049)或不以分子结果为指导的靶向药物(26% 对比 5.2%;P = 0.049)相比,PGT 的客观缓解率为 36%,并改善了 2 年无进展生存= 0.003)。基于一级证据的 PGT、针对融合或在疾病进展之前开始的 PGT 具有最大的临床益处。
- 我们的数据显示,综合分子谱分析提供的 PGT 显着改善了患有高危癌症的儿童的预后。 ClinicalTrials.gov 注册:NCT03336931。
溶瘤腺病毒疗法联合派姆单抗治疗卡介苗无反应的非肌层浸润性膀胱癌:2 期 CORE-001 试验
- Cretostimogene grenadenorepvec 是一种血清型 5 溶瘤腺病毒,旨在选择性地在视网膜母细胞瘤途径发生改变的癌细胞中复制,此前曾在卡介苗 (BCG) 经历的非肌层浸润性膀胱癌中作为单一疗法进行过测试。
- 在这项 2 期研究中,我们评估了膀胱内注射 cretostimogene 和全身性派姆单抗 (pembrolizumab) 对卡介苗无反应的非肌层浸润性膀胱癌伴原位癌 (CIS) 患者的潜在协同疗效。 35 名患者接受了膀胱内 Cretostimogene 联合全身派姆单抗治疗。每周给予诱导 cretostimogene,持续 6 周,然后在第 3、6、9、12 和 18 个月,对保持完全缓解 (CR) 的患者每周进行 3 次维持输注。 3 个月评估中持续存在 CIS/高级别 Ta 的患者有资格进行重新诱导。 Pembrolizumab 的给药时间长达 24 个月。主要终点是 12 个月时的 CR,通过膀胱镜检查、尿细胞学、横断面成像和强制膀胱标测活检进行评估。次要终点包括任何时间的 CR、缓解持续时间、无进展生存期和安全性。
- 12 个月时意向治疗人群的 CR 率为 57.1%(35 例中有 20 例,95% 置信区间 (CI) 40.7-73.5%),达到主要终点。 35 名患者中共有 29 名患者(82.9%,95% CI 70.4-95.3%)在 3 个月时获得 CR。中位随访时间为 26.5 个月,但尚未达到中位缓解持续时间(95% CI 15.7 至未达到)。 24 个月时的 CR 率为 51.4%(35 个月中有 18 个月)(95% CI 34.9-68.0%)。在该试验中,没有患者进展为肌层浸润性膀胱癌。
- 归因于 cretostogene 的不良事件是低度的、自限性的,并且主要限于膀胱相关症状。免疫相关不良反应仅与派姆单抗相关。 35 名患者中共有 5 名 (14.3%) 出现 3 级治疗相关不良反应,全部与派姆单抗相关。没有证据表明存在重叠或协同毒性。
- 膀胱内注射 cretostimogene 和全身帕博利珠单抗的组合显示出持久的疗效。由于其毒性特征与其单一疗法成分相似,这种组合可能会改变 BCG 无反应 CIS 患者的获益风险比。 ClinicalTrials.gov 标识符:NCT04387461。
其它类
单细胞引导产前从人羊水和气管液中提取原代胎儿上皮类器官
- 组织特异性胎儿干细胞的分离和原代类器官的衍生仅限于终止妊娠获得的样本,这阻碍了胎儿发育和先天性疾病的产前研究。因此,需要新的患者特异性体外模型。为此,在怀孕期间分离和扩增胎儿干细胞而不需要组织样本或重新编程将是有利的。羊水 (AF) 是多个发育器官的细胞来源。
- 通过单细胞分析,我们表征了人类 AF 中存在的细胞特性。我们鉴定并分离了胎儿胃肠道、肾脏和肺部来源的活上皮干/祖细胞。培养后,这些细胞形成克隆上皮类器官,表现出小肠、肾小管和肺的特征。 AF 类器官表现出其起源组织的转录组学、蛋白质表达和功能特征。
- 与产前疾病模型相关,我们从先天性膈疝胎儿的 AF 和气管液细胞中衍生出肺类器官,概括了该疾病的一些特征。 AF 类器官是按照与产前干预相一致的时间线衍生的,有可能允许研究针对临床相关发育阶段的胎儿个性化的治疗工具和再生医学策略。
利用功能化纳米孔实时检测 20 种氨基酸并区分病理相关肽
这个还是很强的,为高能量蛋白质精确测序奠定基础
- 氨基酸的精确鉴定和定量对于许多生物应用至关重要。
- 在这里,我们报道了一种带有 N91H 取代的铜 (II) 功能化耻垢分枝杆菌孔蛋白 A (MspA) 纳米孔,与机器学习算法结合使用时,可以直接识别所有 20 种蛋白氨基酸。验证准确率达到99.1%,信号恢复率为30.9%。还证明了在纳摩尔范围内超灵敏定量氨基酸的可行性。
- 此外,该系统还能够使用外肽酶实时分析两种代表性的翻译后修饰 (PTM)、一种非天然氨基酸和十种合成肽,包括与阿尔茨海默病和癌症新抗原相关的临床相关肽。
- 值得注意的是,我们的策略成功地区分出与水解产物仅具有一个氨基酸差异的肽,并提供了推断肽序列的可能性。
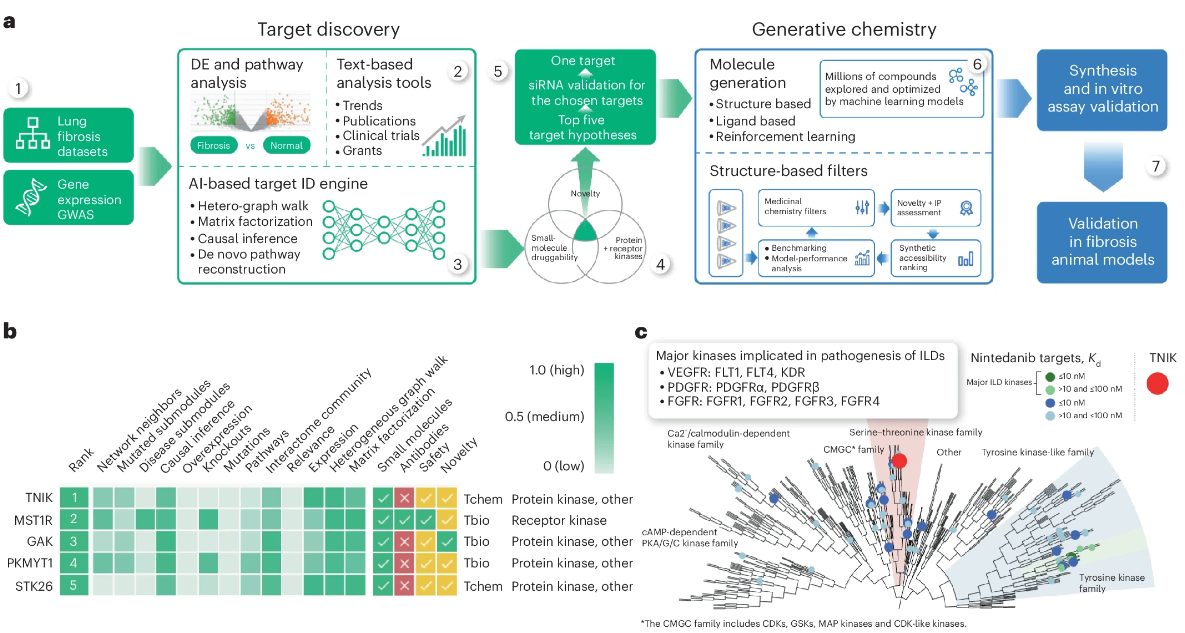
(~ ̄▽ ̄)~ 小分子 TNIK 抑制剂在临床前和临床模型中靶向纤维化
A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models – PubMed. Nat Biotechnol
英科智能医学人工智能有限公司
了解一下方法学
- 特发性肺纤维化(IPF)是一种侵袭性间质性肺疾病,死亡率很高。 IPF 的假定药物靶标未能转化为临床水平的有效疗法。
- 我们使用预测人工智能 (AI) 方法将 TRAF2 和 NCK 相互作用激酶 (TNIK) 确定为抗纤维化靶点。利用人工智能驱动的方法,我们生成了 INS018_055,一种小分子 TNIK 抑制剂,它通过口服、吸入或局部给药在体内不同器官中表现出理想的药物特性和抗纤维化活性。
- INS018_055 除了具有抗纤维化特性外,还具有抗炎作用,已在多项体内研究中得到验证。其安全性、耐受性以及药代动力学在一项涉及 78 名健康参与者的随机、双盲、安慰剂对照 I 期临床试验 (NCT05154240) 中得到验证。在中国进行的另一项 I 期试验 CTR20221542 也证明了类似的安全性和药代动力学特征。
- 这项工作从靶点发现到临床前候选药物提名大约用了 18 个月的时间完成,展示了我们生成式人工智能驱动的药物发现管道的能力。
一种单分子受控释放的光遗传学方法
An optogenetic method for the controlled release of single molecules – PubMed. Nat Methods
德国柏林自由大学化学与生物化学研究所
- 我们开发了一种用于细胞内单分子光遗传学释放的系统。我们通过光裂解蛋白将可溶性跨膜蛋白限制在高尔基体中,并通过短光脉冲释放它们。我们的方法允许以与单分子成像兼容的量将功能蛋白以光剂量依赖性方式递送至细胞质和质膜,从而大大简化了活细胞中任何蛋白质的单分子显微镜检查。
- 我们能够通过将 BK 和 LRRC8/体积调节阴离子通道传递到质膜来重建离子电导。最后,我们能够通过控制释放已被敲除的信号蛋白,在白细胞介素-1 刺激的 T 淋巴母细胞中诱导 NF-kB 信号传导。我们观察到光诱导功能性炎症信号复合物的形成,该复合物仅在活化的细胞中触发核因子 kappa-B 激酶抑制剂的磷酸化。
- 因此,我们开发了一种光遗传学方法,用于在单分子水平上重建和研究细胞功能。
人类基因组长读长测序检测 DNA 甲基化方法的比较
A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes – PubMed (nih.gov). Genome Biol
- 长读长测序可以检测单分子 DNA 中的碱基修饰,例如 CpG 甲基化。最常用的长读长测序方法是Oxford Nanopore Technologies (ONT)开发的纳米孔测序和Pacific Bioscience (PacBio)开发的单分子实时(SMRT)测序。在本研究中,我们系统地比较了长读长测序的 CpG 甲基化检测的性能。
- 我们证明,7179 个纳米孔测序 DNA 样本的 CpG 甲基化检测高度准确,并且与从相同抽血中分离的 132 个氧化亚硫酸氢盐测序 (oxBS) 样本一致。我们引入了 CpG 优质过滤器,进一步提高了纳米孔测序 DNA 中 CpG 甲基化检测的准确性,同时去除了最多 30% 的 CpG。我们评估了不同基因组特征和 CpG 甲基化率的 CpG 甲基化检测的每个位点性能,并演示了最新的 R10.4 流动池化学和碱基识别算法如何改进纳米孔测序的甲基化检测。此外,我们还展示了 50 个 SMRT 测序基因组的甲基化检测与纳米孔测序和 oxBS 的比较。
- 本研究首次对长读长测序方法的 CpG 甲基化检测工具进行了系统比较。我们比较了两种常用的检测大量纳米孔基因组中 CpG 甲基化的计算方法,包括使用最新 R10.4 纳米孔流动池化学测序的样本和 50 个 SMRT 测序样本。我们深入了解每种测序方法的优点和局限性,并为使用长读长测序进行基因组规模修饰碱基检测而设计的工具的标准化和评估提供建议。
RNA Pol II 依赖性转录效率微调 A 到 I 编辑水平
RNA Pol II-dependent transcription efficiency fine-tunes A-to-I editing levels – PubMed. Genome Res
- A-to-I RNA 编辑是一种广泛存在的表观转录组现象,可导致腺苷转化为肌苷,而肌苷主要被细胞机器解释为鸟苷。因此,A-to-I 编辑可以改变剪接或导致转录本重新编码。由于编辑的错误调控可能导致多种人类疾病,因此 A 到 I 编辑需要严格调控脱氨基的程度,特别是在蛋白质编码区域。大部分 A 到 I 编辑是在共转录过程中发生的。因此,我们在转录和前 mRNA 加工的背景下研究了 A 到 I 的编辑调控。
- 我们证明转录的刺激会影响编辑水平。转录因子 MYC 的激活会导致 A 到 I 编辑的上调,特别是在 MYC 激活时受到抑制的转录本中。此外,低前体 mRNA 合成率和低前体 mRNA 表达水平支持高水平的编辑。我们还表明,细胞系统以及小鼠组织中新生前 mRNA 和 mRNA 的编辑水平存在很大差异。编辑水平可以从前 mRNA 到 mRNA 增加或减少,并且可以根据编辑目标和组织的不同而变化,这表明前 mRNA 加工是编辑调控的重要层。多项证据表明差异是在 mRNA 前体剪接过程中出现的。
- 此外,放线菌素 D 对原代神经元细胞的处理和编辑水平分析表明,编辑水平的调节也取决于转录。
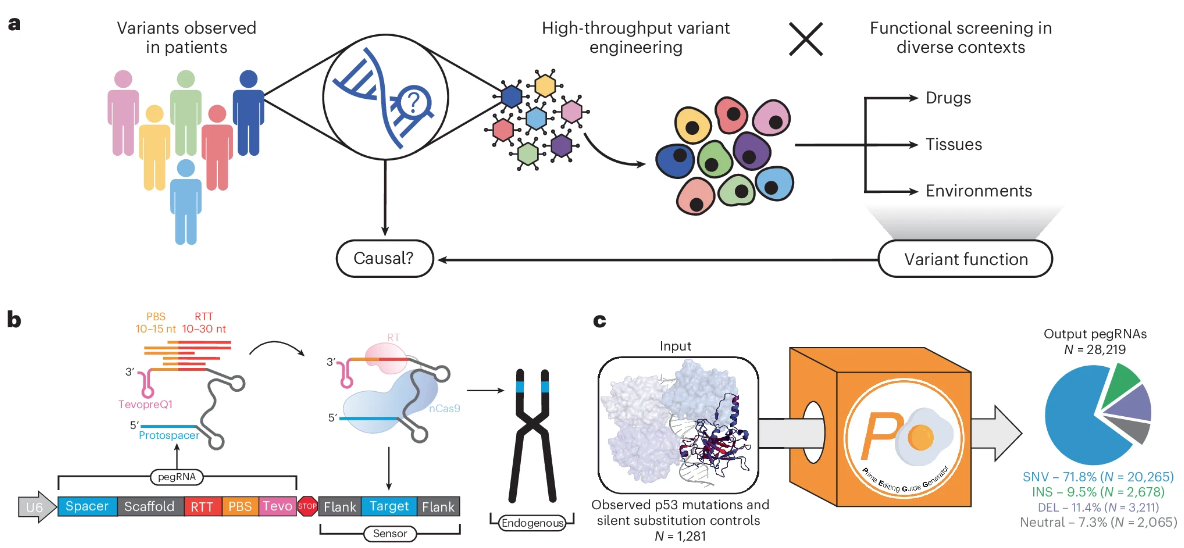
使用prime编辑传感器库对遗传变异进行高通量评估
High-throughput evaluation of genetic variants with prime editing sensor libraries – PubMed (nih.gov). Nat Biotechnol. full html
- 肿瘤基因组通常包含一系列复杂的单核苷酸改变和染色体重排,可能会扰乱蛋白质功能。 Prime 编辑已被应用于安装和评估遗传变异,但以前的方法受到 Prime 编辑引导 RNA 的可变效率的限制。
- 在这里,我们提出了一种高通量prime编辑传感器策略,将prime编辑引导RNA与其同源靶位点的合成版本结合起来,以定量评估内源遗传变异的功能影响。
- 我们筛选了超过 1,000 种与癌症相关的 TP53 内源性变体(癌症中最常见的突变基因),以确定以不同机制影响 p53 功能的等位基因。我们发现某些内源 TP53 变体,特别是 p53 寡聚化结构域中的变体,在外源过表达系统中表现出相反的表型。
- 我们的结果强调了基因剂量在塑造天然蛋白质化学计量和蛋白质-蛋白质相互作用方面的生理重要性,并建立了大规模研究内源序列背景下的遗传变异的框架。
非编码CRISPRI屏幕的多中心集成分析
Multicenter integrated analysis of noncoding CRISPRi screens – PubMed (nih.gov)
- ENCODE 联盟在注释非编码顺式调控元件 (CRE) 方面所做的努力增进了我们对基因调控景观的理解。汇集的非编码 CRISPR 筛选提供了一种研究顺式调控机制的系统方法。
- ENCODE4 功能表征中心对人类细胞系进行了 108 次筛选,在基因组的 24.85 兆碱基中包含超过 540,000 个扰动。利用 K562 细胞中 332 个经功能确认的 CRE 基因链接,我们制定了利用 CRISPR 干扰 (CRISPRi) 筛选内源非编码元件的指南,包括准确检测表现出可变且通常较低的转录效应的 CRE。
- 通过对五种筛选分析工具进行基准测试,我们发现 CASA 产生最保守的 CRE 调用,并且对低特异性单向导 RNA 的伪影具有鲁棒性。我们发现了转录区域中 CRISPRi 的微妙 DNA 链偏差,这对筛选设计和分析具有影响。
- 我们共同提供可访问的数据资源、预先设计的单引导 RNA(用于使用 CRISPRi 靶向 3,275,697 个编码筛选候选 CRE)以及筛选指南,以加速非编码基因组的功能表征。
DART.2:具有千倍细胞特异性的双向突触药理学
DART.2: bidirectional synaptic pharmacology with thousandfold cellular specificity – PubMed. Nat Methods
- 精准药理学旨在操纵复杂组织内的特定细胞相互作用。
- 为了实现这一目标,我们推出了第二代细胞特异性药理学技术 DART.2(drug acutely restricted by tethering)。核心进步是优化的细胞特异性(15 分钟内高达 3,000 倍),甚至能够靶向递送致癫痫药物,而不会产生脱靶效应。此外,我们引入全脑给药方法作为局部插管和示踪试剂的替代方案,用于全脑剂量定量。
- 我们描述了四种药物——两种拮抗兴奋性和抑制性突触后受体,另两种以变构方式增强这些受体。它们的多功能性在小鼠大脑的多个区域中得到体现,包括小脑、纹状体、视觉皮层和视网膜。最后,在腹侧被盖区,我们发现阻断多巴胺神经元的抑制性输入会加速运动,这与之前的光遗传学和药理学发现形成鲜明对比。
- 除了实现化学突触的双向扰动之外,这些试剂还提供基因定义的突触后细胞和神经递质定义的突触前伙伴之间的交叉精度。
分支化学修饰的聚腺苷酸尾增强 mRNA 的翻译能力
Branched chemically modified poly(A) tails enhance the translation capacity of mRNA – PubMed. Nat Biotechonol
美国麻省理工学院化学系
- 尽管信使 RNA (mRNA) 已被证明作为疫苗是有效的,但其作为一般治疗方式的潜力因其不稳定性和低翻译能力而受到限制。
- 为了增加 mRNA 蛋白质表达的持续时间和水平,我们设计并合成了具有多个合成 Poly(A) 尾的拓扑和化学修饰的 mRNA。
- 在这里,我们证明,在细胞转染后 24 至 72 小时,优化的多尾 mRNA 产生的发光信号比对照 mRNA 高约 4.7-19.5 倍,并且与体内对照的信号相比,14 天可检测到的信号小于 7 天。我们进一步以最小的 mRNA 剂量实现了小鼠肝脏中临床相关基因 Pcsk9 和 Angptl3 的高效多重基因组编辑。
- 总而言之,这些结果提供了一种合成带帽分支 mRNA 的通用方法,其翻译能力显着增强。
多模态细胞分割挑战:迈向通用解决方案
The multimodality cell segmentation challenge: toward universal solutions – PubMed. Nat Methods
- 细胞分割是显微图像中定量单细胞分析的关键步骤。现有的细胞分割方法通常是针对特定模式量身定制的,或者需要手动干预来指定不同实验设置中的超参数。
- 在这里,我们提出了一个多模态细胞分割基准,包括来自 50 多个不同生物实验的 1,500 多个标记图像。
- 顶级参与者开发了一种基于 Transformer 的深度学习算法,该算法不仅超越了现有方法,而且还可以应用于跨成像平台和组织类型的各种显微图像,而无需手动调整参数。
- 该基准和改进的算法为显微成像中更准确和更通用的细胞分析提供了有希望的途径。
临床基因组测序对全球疑似罕见遗传病人群的影响
The impact of clinical genome sequencing in a global population with suspected rare genetic disease – PubMed. Am J Hum Genet
- 越来越多的证据表明临床基因组测序 (cGS) 对疑似罕见遗传病 (RGD) 患者的价值,但 cGS 在来自高收入国家 (HIC) 和低收入国家的不同人群中的表现和对临床护理的影响和中等收入国家(LMIC)尚未进行调查。 iHope 计划是一项慈善 cGS 计划,在 8 个国家建立了一个由 24 个临床中心组成的网络,通过该网络向有 RGD 体征或症状且分子检测机会有限的个人提供 cGS。
- 2016 年 6 月至 2021 年 9 月期间,共有 1,004 名具有不同祖先背景(51.8% 非多数欧洲人)的个体(中位年龄 6.5 岁;53.5% 男性)接受了评估。cGS 的诊断率为 41.4% (416/1,004) ,与 HIC 站点相比,来自 LMIC 站点的个体获得阳性检测结果的可能性高出 1.7 倍(LMIC 56.5% [195/345] vs. HIC 33.5% [221/659],OR 2.6,95% CI 1.9-3.4, p < 0.0001)。 76.9% (514/668) 的个体诊断评估发生变化。
- 据报道,41.4% (285/694) 的个体改变了管理方式,包括专业转诊、影像和检测、治疗干预和姑息治疗,而当进行遗传咨询和避免额外治疗时,这一比例增加至 69.2% (480/694)。测试也包括在内。来自 LMIC 地点的个体与 HIC 同行一样有可能经历诊断评估的变化(OR 6.1,95% CI 1.1-∞,p = 0.05)和管理变化(OR 0.9,95% CI 0.5-1.3,p = 0.49)。
- 增加基因组检测的机会可能有助于诊断的公平性和减少全球医疗保健差距。
年龄相关和代谢相关肠道微生物特征与心血管疾病风险
- 对肠道微生物组与代谢和衰老关联的深入了解,对于制定促进健康长寿的干预措施至关重要。
- 在一个由10,207名年龄在40至93岁之间的个体组成的发现队列中,我们使用21个代谢参数将个体分类为五个集群,称为代谢多发病集群(multimorbidity clusters, MCs),代表不同的代谢亚表型。
- 与分类为代谢健康的集群(MC1)相比,分类为“肥胖相关混合”(MC4)和“高血糖”(MC5)的集群在11.1年的心血管疾病(CVD)风险中分别增加了75%(多变量调整HR:1.75,95% CI :1.43-2.14)和117%(2.17,1.72-2.74)。这些关联在第二个由9,061名个体组成的队列中得到了验证,该队列的随访时间为10.0年。
- 基于发现队列中4,491个粪便宏基因组的分析,我们发现肠道微生物组成与MCs和年龄均相关。接下来,使用55种特定年龄的微生物物种来捕捉生物年龄,我们开发了一个肠道微生物年龄(MA)指标,该指标在四个包含4,425个宏基因组样本的外部队列中得到了验证。在60岁或以上的个体中,与MC1、MC2或MC3相比,MC4或MC5相关的CVD风险在高MA个体中加剧,但在低MA个体中减弱,独立于年龄、性别和其他生活方式及饮食因素。
- 这种模式表明,在代谢不健康的老年人中,较年轻的MA似乎可以抵消由代谢功能障碍导致的CVD风险,暗示了MA在心血管健康中的调节作用。
机器学习/组学类
跨癌症类型联合免疫检查点阻断的肠道微生物特征
A gut microbial signature for combination immune checkpoint blockade across cancer types – PubMed. Nat Methods
- 针对程序性细胞死亡蛋白 1 (PD-1) 和细胞毒性 T 淋巴细胞蛋白 4 (CTLA-4) 的免疫检查点阻断 (ICB) 可以在多种癌症中诱导显着但不可预测的反应。研究表明,癌症患者的肠道微生物群组成与 ICB 的临床反应之间存在关联;然而,定义跨群体的基于微生物组的生物标志物一直具有挑战性。这可能与之前将微生物群量化为物种(或更高分类等级)丰度的努力有关,而微生物功能通常是菌株特异性的。
- 在这里,我们对来自独特的、注释丰富的 2 期试验队列的基线粪便样本进行了深度鸟枪法宏基因组测序,该队列的患者患有多种罕见癌症,接受联合 ICB 治疗(n = 106 发现队列)。
- 我们证明,相对于使用物种等级量化或综合预处理临床因素构建的模型,菌株解析的微生物丰度改善了 ICB 反应和 12 个月无进展生存的机器学习预测。通过对另外六项可比研究(n = 364 验证队列)的肠道宏基因组进行荟萃分析,我们发现菌株反应特征的跨癌症(和跨国)有效性,但前提是训练和测试队列使用一致ICB 方案(抗 PD-1 单一疗法或抗 PD-1 加抗 CTLA-4 联合疗法)。
- 这表明肠道微生物组诊断或治疗的未来发展应根据 ICB 治疗方案而不是根据癌症类型进行定制。
(~ ̄▽ ̄)~ 表征数据集不平衡对单细胞数据集成的影响
Characterizing the impacts of dataset imbalance on single-cell data integration – PubMed. Nat Biotechnol
- 用于整合来自多个样本和条件的单细胞转录组数据的计算方法通常不能解释不同数据集中测量的细胞类型的不平衡。在这项研究中,我们研究了样品中存在的细胞类型、每种细胞类型的细胞数量以及样品中细胞类型比例的差异如何影响整合后的下游分析。
- Iniquitate 管道在扰乱数据集之间的不平衡程度后评估集成结果的稳健性。在 2,600 次整合实验中对五种最先进的单细胞 RNA 测序整合技术进行的基准测试表明,样本不平衡对下游分析和整合结果的生物学解释具有重大影响。不平衡扰动导致无监督聚类、细胞类型分类、差异表达和标记基因注释、查询到参考映射和轨迹推断方面出现统计上显着的变化。我们通过新引入的属性——聚合细胞类型支持和最小细胞类型中心距离来量化不平衡的影响。
- 为了更好地表征和减轻不平衡的影响,我们为集成方法用户引入了平衡聚类指标和不平衡集成指南。
scAbsolute:测量单细胞倍性和复制状态
scAbsolute: measuring single-cell ploidy and replication status – PubMed. Genome Biol
- 癌细胞经常表现出 DNA 拷贝数畸变,并且其倍性差异很大。正确估计单细胞基因组的倍性对于下游分析至关重要。
- scAbsolute 仅基于单细胞 DNA 测序信息,即可实现对单细胞倍性和复制状态(包括全基因组重复)的准确且公正的测量。
- 我们使用实验细胞多重、FUCCI 细胞周期表达系统和针对最先进方法的基准来展示 scAbsolute 的功能。
- scAbsolute 为跨不同技术的单细胞 DNA 测序分析提供了坚实的基础,并有可能改进许多下游分析。
从 scRNA-seq 数据预测肿瘤反应性 T 细胞受体用于个性化 T 细胞治疗
Prediction of tumor-reactive T cell receptors from scRNA-seq data for personalized T cell therapy – PubMed. Nat Biotechnol
- 识别源自患者的肿瘤反应性 T 细胞受体 (TCR) 作为个性化转基因 T 细胞疗法的基础仍然是一项耗时且成本密集的工作。目前识别肿瘤反应性 TCR 的方法是通过分析肿瘤突变来预测 T 细胞激活(新)抗原,并利用这些抗原来富集肿瘤浸润淋巴细胞 (TIL) 培养物或验证个体 TCR 用于转基因自体疗法。
- 在这里,我们结合高通量 TCR 克隆和反应性验证来训练 predicTCR,这是一种机器学习分类器,可基于单 TIL RNA 测序以与抗原无关的方式识别个体肿瘤反应性 TIL。 PredicTCR 比以前基于基因集富集的方法更好地识别来自不同癌症的 TIL 中的肿瘤反应性 TCR,将特异性和灵敏度(几何平均值)从 0.38 提高到 0.74。
- 通过在几天内预测肿瘤反应性 TCR,可以优先考虑 TCR 克隆型,以加速个性化 T 细胞疗法的制造。
- Methods: Data were imported in Python (v3.9.16) using pandas (v2.0.2) for preprocessing before training with xgboost (v1.7.4). Due to the scRNA data having many dropouts, we performed hyperparameter tuning before feature selection. The XGBoost hyperparameters ‘colsample_bytree’, ‘gamma’, ‘learning_rate’, ‘max_delta_step’, ‘max_depth’, ‘min_child_weight’, ‘n_estimators’, ‘alpha’, ‘lambda’, ‘scale_pos_weight’ and ‘subsample’ were tuned by Bayesian optimization using scikit-optimize (v0.9.0)
对人类癌症突变和转录景观的联合分析揭示了癌症进化过程中的关键扰动
美国马萨诸塞州波士顿布莱根妇女医院和哈佛医学院基因莱免疫学和炎症研究所
- 肿瘤能够获得新的能力,包括与不良临床结果相关的耐药性和转移等特征。单细胞技术使研究突变和转录组谱成为可能,但由于大多数研究都是在模型系统上进行的,因此对人类患者癌症进化知之甚少。因此,更好地了解癌症进化可能对治疗策略产生重要影响。
- 在这里,我们通过共同考虑从 49 个肺癌样本和 51 个慢性粒细胞白血病样本的肿瘤活检中获得的单细胞的突变和转录组谱来分析癌症进化和克隆选择。比较这两个图谱,我们发现每个克隆都与首选转录状态相关。对于转移和耐药性,我们发现影响相关基因的突变数量随着克隆的进化而增加,而基因表达谱的变化有限。令人惊讶的是,我们发现随着克隆获得耐药性,影响配体-受体与肿瘤微环境相互作用的突变经常出现。
- 我们的结果表明肺癌和慢性粒细胞白血病保持着较高的克隆和转录多样性,并且我们几乎没有发现支持克隆清除的证据。这表明对于这些癌症来说,仅基于生长速度的选择不太可能成为癌症进化过程中的主导驱动力。
使用空间解析转录组数据对空间聚类方法进行基准测试
Benchmarking spatial clustering methods with spatially resolved transcriptomics data – PubMed (nih.gov). Nat Methods
中国科学院计算技术研究所普适计算系统研究中心
- 空间聚类与单细胞聚类相似,利用空间解析转录组学(SRT)数据将组织生理学研究的范围从细胞质心扩展到结构质心。近年来计算方法取得了显着的发展,但仍然缺乏全面的基准研究。
- 在这里,我们提出了对 34 个 SRT 数据(7 个数据集)的 13 种计算方法的基准研究。根据准确性、空间连续性、标记基因检测、可扩展性和鲁棒性来评估性能。
- 我们发现现有方法在性能和功能方面是互补的,并且我们提供了针对给定场景选择适当方法的指导。在测试另外 22 个具有挑战性的数据集时,我们发现了识别不连续空间域的挑战以及现有方法的局限性,突出了它们在处理最近的大规模任务方面的不足。此外,我们利用 145 个模拟数据,检查了这些方法针对四个不同因素的稳健性,并评估了预处理和后处理方法的影响。
- 我们的研究利用 SRT 数据对现有空间聚类方法进行了全面评估,为这个快速发展的领域的未来发展铺平了道路。
人工智能辅助对放射科医生的影响的异质性和预测因素
Heterogeneity and predictors of the effects of AI assistance on radiologists – PubMed (nih.gov). Nat Med
- 人工智能(AI)在医学图像判读中的集成需要临床医生和人工智能算法之间的有效协作。尽管之前的研究证明了人工智能辅助在提高临床医生整体绩效方面的潜力,但对临床医生的个人影响仍不清楚。
- 这项大规模研究检查了人工智能辅助对 140 名放射科医生执行 15 项胸部 X 光诊断任务的异质性影响,并确定了这些影响的预测因素。令人惊讶的是,传统的基于经验的因素,例如多年的经验、专业知识和对人工智能工具的熟悉程度,无法可靠地预测人工智能援助的影响。此外,表现较差的放射科医生并没有始终从人工智能的帮助中受益更多,这挑战了普遍的假设。
- 相反,我们发现人工智能错误的发生强烈影响治疗结果,不准确的人工智能预测会对放射科医生对所有病理的总体表现以及所调查的一半的个体病理的表现产生不利影响。
- 我们的研究结果强调了临床医生与人工智能协作的个性化方法的重要性以及准确的人工智能模型的重要性。通过了解影响人工智能辅助有效性的因素,这项研究为人工智能的针对性实施提供了宝贵的见解,从而在临床实践中为个体临床医生带来最大的利益。
(~ ̄▽ ̄)~ 系列研究:计算病理学的视觉语言基础模型
A visual-language foundation model for computational pathology – PubMed (nih.gov). Nat Med
波士顿哈佛医学院布莱根妇女医院病理科Faisal Mahmood团队
- 数字病理学的加速采用和深度学习的进步使得能够为各种疾病和患者群体的各种病理学任务开发强大的模型。然而,由于医学领域的标签稀缺,模型训练通常很困难,并且模型的使用受到训练的特定任务和疾病的限制。此外,大多数组织病理学模型仅利用图像数据,这与人类相互教导和推理组织病理学实体的方式形成鲜明对比。
- 我们引入了来自CONtrastive learning from Captions for Histopathology (CONCH) ,这是一种视觉语言基础模型,使用多种来源的组织病理学图像、生物医学文本,尤其是通过与任务无关的预训练超过 117 万个图像字幕对而开发。
- CONCH 根据一套 14 个不同的基准进行评估,可以转移到涉及组织病理学图像和/或文本的广泛下游任务,在组织学图像分类、分割、字幕和文本到文本转换方面实现最先进的性能-图像和图像到文本的检索。
- CONCH 代表了组织病理学并发视觉语言预训练系统的重大飞跃,有可能直接促进各种基于机器学习的工作流程,需要最少或不需要进一步的监督微调。
(~ ̄▽ ̄)~ 系列研究:计算病理学的通用基础模型
Towards a general-purpose foundation model for computational pathology – PubMed (nih.gov). Nat Med
波士顿哈佛医学院布莱根妇女医院病理科Faisal Mahmood团队
了解计算病理学的一个不错的入门文章
- 组织图像的定量评估对于计算病理学 (CPath) 任务至关重要,需要从全幻灯片图像 (WSI) 中客观表征组织病理学实体。 WSI 的高分辨率和形态特征的可变性提出了重大挑战,使高性能应用的大规模数据注释变得复杂。为了应对这一挑战,当前的工作提出通过自然图像数据集的迁移学习或对公开的组织病理学数据集的自我监督学习来使用预训练的图像编码器,但尚未在不同的组织类型中大规模地广泛开发和评估。
- 我们推出 UNI,一种通用的病理学自我监督模型,使用来自 20 个主要组织类型的 100,000 多个诊断 H&E 染色 WSI(>77 TB 数据)的 1 亿多张图像进行预训练。该模型在 34 个具有不同诊断难度的代表性 CPath 任务上进行了评估。除了超越以前最先进的模型之外,我们还展示了 CPath 中的新建模功能,例如与分辨率无关的组织分类、使用少样本类原型的幻灯片分类以及对OncoTree 分类系统中多达 108 种癌症类型进行分类的疾病亚型概括。
- UNI 在预训练数据和下游评估方面在 CPath 中大规模推进无监督表示学习,从而实现数据高效的人工智能模型,该模型可以泛化并转移到解剖病理学中的各种具有诊断挑战性的任务和临床工作流程。
scPROTEIN:用于单细胞蛋白质组嵌入的多功能深度图对比学习框架
南开大学人工智能学院、腾讯AI实验室[健康护理]
- 单细胞蛋白质组测序技术揭示了细胞中蛋白质-蛋白质相互作用、翻译后修饰和蛋白质形式动力学。然而,肽定量的不确定性估计、数据缺失、批次效应和高噪声阻碍了单细胞蛋白质组数据的分析。共同解决这一系列错综复杂的问题非常重要,但现有的针对单细胞转录组的方法无法完全解决这项任务。
- 在这里,我们提出了一个专为单细胞蛋白质组数据分析而设计的通用框架,称为 scPROTEIN,它由基于多任务异方差回归模型的肽不确定性估计和基于图对比学习的细胞嵌入生成组成。
- scPROTEIN 可以估计肽定量的不确定性、对蛋白质数据进行降噪、消除批次效应并在统一框架中编码单细胞蛋白质组特异性嵌入。
- 我们证明 scPROTEIN 对于细胞聚类、批量校正、细胞类型注释、临床分析和空间解析蛋白质组数据探索是有效的。
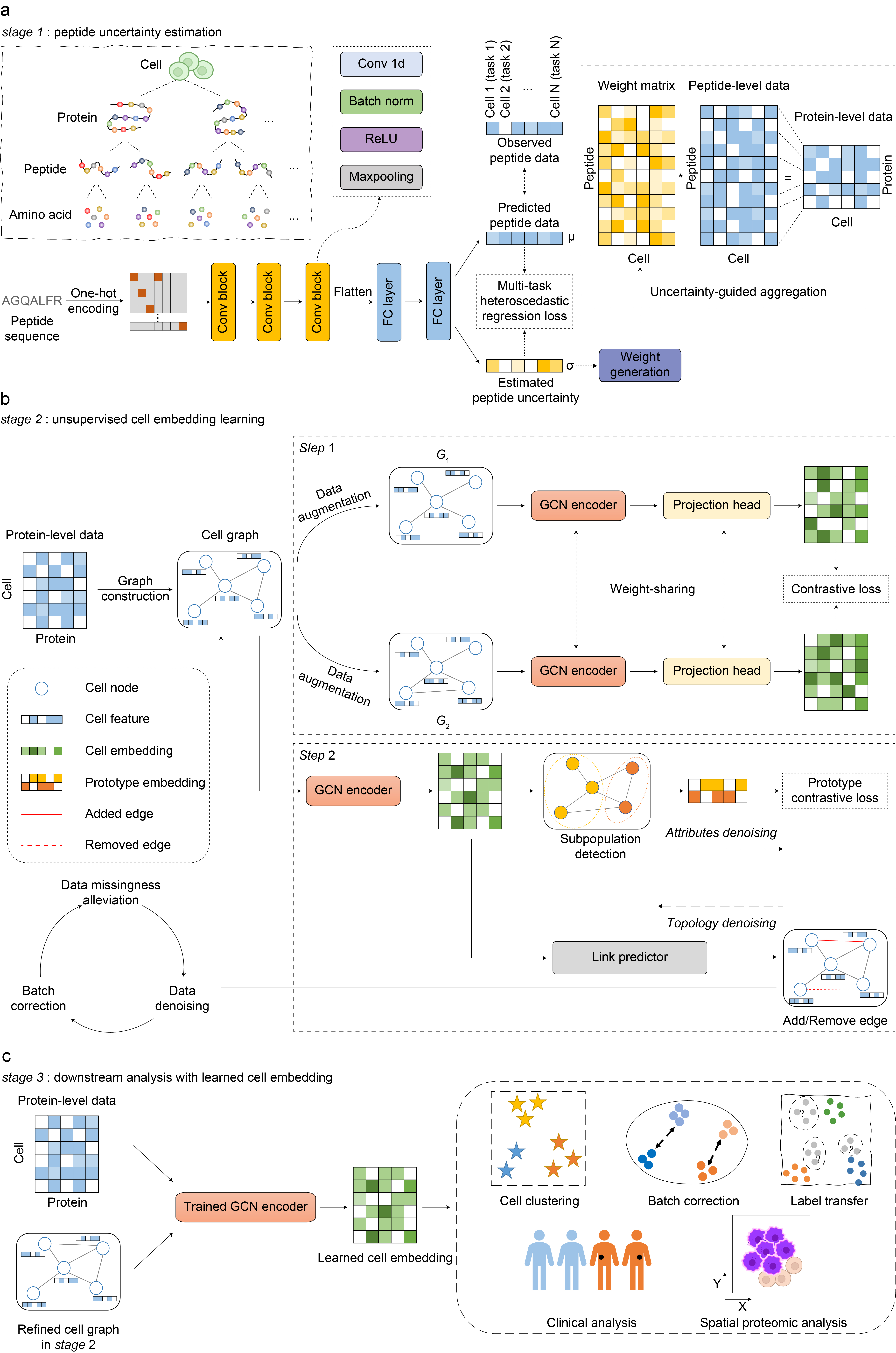
DANCE:用于单细胞分析的深度学习库和基准平台
DANCE: a deep learning library and benchmark platform for single-cell analysis – PubMed. Genome Biol
- DANCE – python 是第一个标准、通用且可扩展的基准平台,用于访问和评估众多单细胞分析任务的基准数据集范围内的计算方法。
- 目前,DANCE 支持 3 个模块和 8 个流行任务,在 21 个基准数据集上使用 32 种最先进的方法。人们可以通过最少的努力(例如仅使用一个命令行)轻松地在主要基准数据集上重现支持的算法的结果。
- 此外,DANCE 还为研究人员提供了一个由深度学习架构和工具组成的生态系统,以促进他们自己的模型开发。
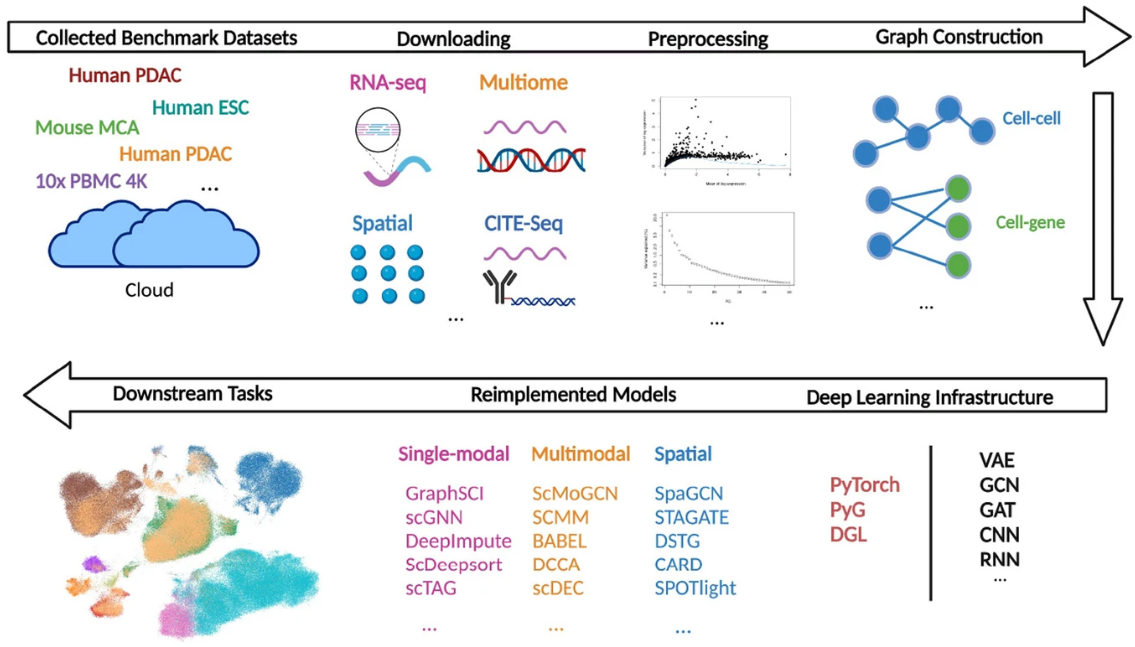
SpatialData:空间组学的开放通用数据框架
SpatialData: an open and universal data framework for spatial omics – PubMed. Nat Methods. full html
- 空间分辨组学技术正在改变我们对生物组织的理解。然而,由于数据量大、数据类型的异质性以及缺乏灵活的空间感知数据结构,单模态和多模态空间组学数据集的处理仍然是一个挑战。
- 在这里,我们介绍 SpatialData框架,它建立了统一且可扩展的多平台文件格式、大于内存数据的惰性表示、转换和与公共坐标系的对齐。
- SpatialData 有助于空间注释以及跨模式聚合和分析,其实用性在多个小插图的背景下进行了说明,包括对多模式 Xenium 和 Visium 乳腺癌研究的综合分析。
SQANTI3:管理长读转录组以准确识别已知和新型亚型
SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms – PubMed. Nat Methods. full html
- SQANTI3 是一款专为对通过第三代测序技术获得的长读转录本模型进行质量控制、管理和注释而设计的工具。
- SQANTI3 利用其注释框架计算转录模型、连接点和转录末端的质量描述符。有了这些信息,就可以识别潜在的伪影并用可靠的序列替换。
- 此外,集成的功能注释功能可以实现后续的功能同转录组分析。
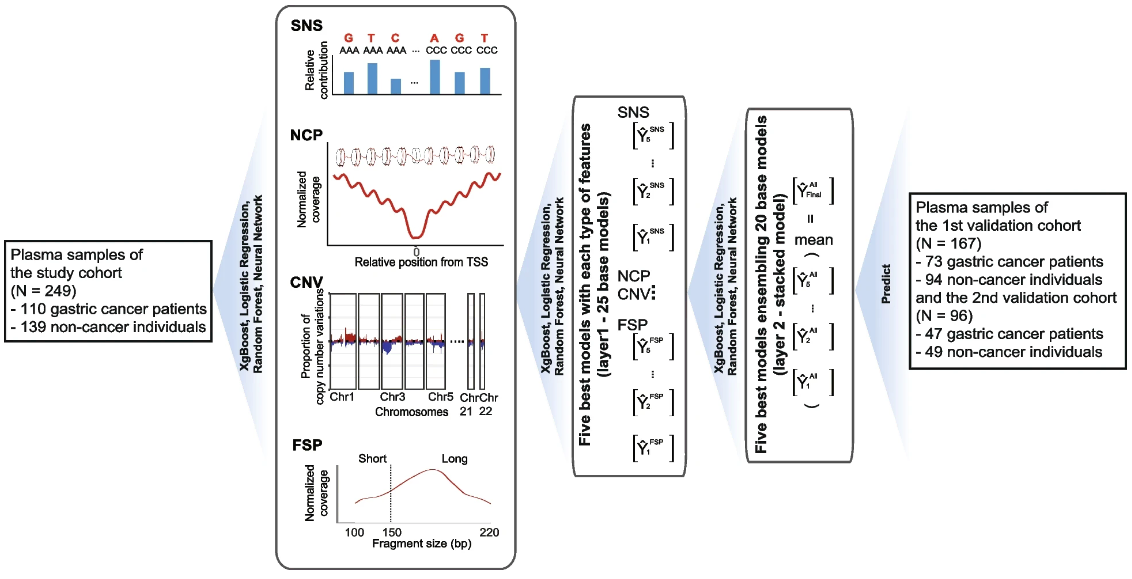
基于多维 cfDNA 的液体活检用于胃癌的灵敏早期检测
Multi-dimensional cell-free DNA-based liquid biopsy for sensitive early detection of gastric cancer – PubMed. Genome Med
中国科学院杭州医学研究所、浙江省肿瘤医院胃外科
- 胃癌是第五大常见癌症类型。大多数患者确诊时已属晚期,预后较差。非常需要一种用于检测早期胃癌的非侵入性检测方法,以降低相关死亡率。
- 我们收集了 110 名 I-II 期胃癌患者和 139 名非癌症个体组成的前瞻性研究队列。我们对血浆样本进行了全基因组测序,并分析了四种类型的游离 DNA (cfDNA) 特征、片段大小模式、拷贝数变异、核小体覆盖模式和单核苷酸取代。利用这些差异特征,我们开发了一个集成模型来检测胃癌信号。此外,我们在由 73 名胃癌患者和 94 名非癌症个体组成的内部第一验证队列以及由 47 名胃癌患者和 49 名非癌症个体组成的独立第二验证队列中验证了该检测方法。此外,我们通过蒙特卡罗模拟在假设的 100,000 名筛查人群中评估了该检测方法。
- 我们基于 cfDNA 的检测可以区分早期胃癌和非癌症,研究队列中的 AUROC 为 0.962 (95% CI: 0.942-0.982),第一个队列中的 AUROC 为 0.972 (95% CI: 0.953-0.992)验证队列中的值为 0.937 (95% CI: 0.890-0.983)。该模型在研究队列中的特异性达到 92.1% (128/139),敏感性达到 88.2% (97/110)。在第一个验证队列中,91.5% (86/94) 的非癌症个体和 91.8% (67/73) 的胃癌患者被正确识别。在第二个验证队列中,89.8% (44/49) 的非癌症个体和 87.2% (41/47) 的胃癌患者被准确分类。
- 我们引入了一种使用多维 cfDNA 特征的液体活检检测方法,可以准确地将早期胃癌与非癌症疾病区分开来。作为一种具有成本效益的非侵入性方法,它可以为胃癌的早期检测提供全民受益。
用于转录本鉴定和定量的长读长 RNA-seq 方法的系统评估
Systematic assessment of long-read RNA-seq methods for transcript identification and quantification – PubMed. Nat Methods
西班牙国家研究委员会 (CSIC) 综合系统生物学研究所
- 长读长 RNA-Seq 基因组注释评估项目联盟的成立是为了评估长读长转录组分析方法的有效性。该联盟使用不同的协议和测序平台,从互补 DNA 和直接 RNA 数据集中生成了超过 4.27 亿条长读序列,涵盖人类、小鼠和海牛物种。开发人员利用这些数据来解决转录亚型检测、定量和从头转录检测方面的挑战。
- 研究表明,具有更长、更准确序列的文库比具有增加读取深度的文库产生更准确的转录本,而更大的读取深度则提高了定量准确性。在注释良好的基因组中,基于参考序列的工具表现出最佳性能。当旨在检测罕见和新颖的转录本或使用无参考方法时,建议结合额外的正交数据和重复样本。
- 这项合作研究为当前实践提供了基准,并为转录组分析的未来方法开发提供了方向。
CelFiE-ISH:单分子 DNA 甲基化单倍型多细胞类型反卷积的概率模型
- 反卷积方法通过对包括血液和组织在内的混合样本的批量测量来推断定量细胞类型估计。 DNA 甲基化测序可测量每次读取的多个 CpG,但现有的反卷积方法很少利用这种within-read信息。
- 我们开发了 CelFiE-ISH,它扩展了现有方法 (CelFiE) 以使用within-read单倍型信息。
- CelFiE-ISH 优于 CelFiE 和其他现有方法,对稀有细胞类型的检测精度提高了 30%,检测灵敏度更高。
- 我们还证明了标记选择和定制标记对于单倍型感知方法的重要性。虽然我们在这里使用黄金标准的短读长测序数据,但单倍型感知方法将非常适合长读长测序。
单样本肿瘤亚克隆重建的众包基准测试
Crowd-sourced benchmarking of single-sample tumor subclonal reconstruction – PubMed. Nat Biotechnol
- 亚克隆重建算法使用大量 DNA 测序数据来量化肿瘤进化的参数,从而可以评估癌症如何发生、进展和对选择压力的反应。
- 我们发起了 ICGC-TCGA(国际癌症基因组联盟 – 癌症基因组图谱)DREAM 体细胞突变调用肿瘤异质性和进化挑战赛,以对现有亚克隆重建算法进行基准测试。
- 这项为期 7 年的社区努力使用云计算对 51 个模拟肿瘤的 31 种亚克隆重建算法进行了基准测试。对 7 个独立任务的算法进行了评分,总共运行了 12,061 次。算法选择对性能的影响远远大于肿瘤特征,但纯度调整的读取深度、拷贝数状态和读取可映射性与大多数算法在大多数任务上的性能相关。没有任何一种算法能够在所有七项任务中表现最佳,并且现有的集成策略无法超越最佳的单独方法,这凸显了关键的研究需求。
- 所有容器化方法、评估代码和数据集均可用于支持进一步评估亚克隆重建准确性的决定因素以及开发改进方法以了解肿瘤进化。
基于单细胞成像的空间分辨转录组学中的基因计数标准化
Gene count normalization in single-cell imaging-based spatially resolved transcriptomics – PubMed. Genome Biol
- 基于成像的空间解析转录组学 (im-SRT) 技术的最新进展现在能够对目标基因及其在固定组织中的位置进行高通量分析。通常需要对基因表达数据进行标准化,以考虑可能混淆潜在生物信号的技术因素。
- 在这里,我们研究了不同基因计数标准化方法与不同目标基因组在 im-SRT 数据分析和解释中的潜在影响。使用过度代表特定组织区域或细胞类型中表达的基因的不同模拟基因组,我们演示了基于每个细胞检测到的基因计数的标准化方法如何以区域或细胞类型特异性的方式差异影响标准化基因表达量。
- 我们表明,这些归一化诱导的效应可能会降低下游分析的可靠性,包括差异基因表达、基因倍数变化和空间可变基因分析,与从更能代表基因组的结果相比,引入假阳性和假阴性结果。组织组成细胞类型的基因表达。如果使用不使用检测到的基因计数来调整基因表达量的标准化方法(例如细胞体积或细胞面积标准化),则不会观察到这些效应。
- 我们建议在可行的情况下使用基于非基因计数的标准化方法,并在必要时在使用基于基因计数的标准化方法之前评估基因组的代表性。总的来说,我们警告标准化方法和基因组的选择可能会影响 im-SRT 数据的生物学解释。
BRAKER3:使用 RNA-seq 和 GeneMark-ETP、AUGUSTUS 和 TSEBRA 的蛋白质证据进行全自动基因组注释
- 基因预测长期以来一直是生物信息学研究的活跃领域。尽管如此,大型真核基因组中的基因预测仍然是一个必须通过新算法来解决的挑战。转录组和蛋白质组提供的证据的数量和重要性因基因组、基因之间甚至单个基因而异。需要能够应对这种数据异构性的用户友好且准确的注释管道。之前开发的注释管道 BRAKER1 和 BRAKER2 分别使用 RNA-seq 或蛋白质数据,但不能同时使用两者。最近发布的 GeneMark-ETP 集成了所有三种数据类型,进一步显着提高了性能。
- 我们在此介绍基于 GeneMark-ETP 和 AUGUSTUS 构建的 BRAKER3 流程,并使用 TSEBRA 组合器进一步提高准确性。 BRAKER3 使用短读长 RNA-seq 和大型蛋白质数据库以及针对目标基因组迭代学习的统计模型来注释真核基因组中的蛋白质编码基因。我们在假设目标物种蛋白质组与可用蛋白质组的相关性水平下,对 11 个物种的基因组的新管道进行了基准测试。
- BRAKER3 优于 BRAKER1 和 BRAKER2。平均转录水平 F1 分数平均增加约 20 个百分点,而对于基因组庞大且复杂的物种来说,这种差异最为明显。 BRAKER3 的性能还优于其他现有工具,如 MAKER2、Funannotate 和 FINDER。
- BRAKER3 的代码可在 GitHub 上获取,并作为可立即运行的 Docker 容器,用于通过 Docker 或 Singularity 执行。总体而言,BRAKER3 是一种准确、易于使用的真核基因组注释工具。
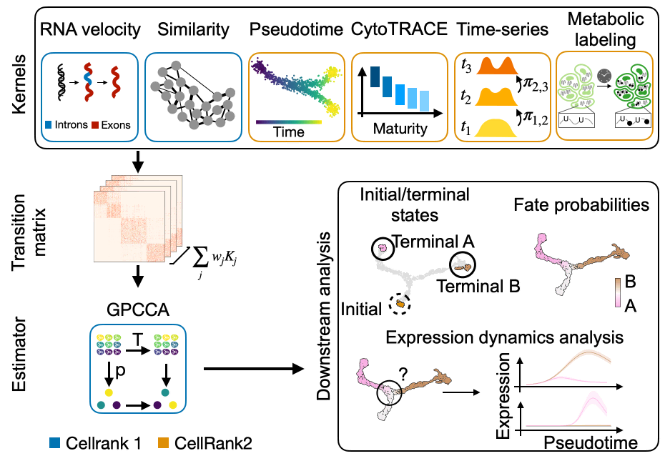
CellRank 2:多视图单细胞数据中的统一命运映射
CellRank 2: unified fate mapping in multiview single-cell data – PubMed. Nat Methods
- 单细胞 RNA 测序使我们能够使用表达相似性或 RNA 速度来模拟细胞状态动态和命运决定,以重建状态变化轨迹;然而,轨迹推断不包含有价值的时间点信息或利用额外的模式,而解决这些不同数据视图的方法不能组合或不能扩展。
- 在这里,我们介绍 CellRank 2,这是一个多功能且可扩展的框架,用于以统一的方式使用多达数百万个细胞的多视图单细胞数据来研究细胞命运。
- CellRank 2 一致地恢复人类造血和内胚层发育的各种数据模式的终末状态和命运概率。我们的框架还允许组合实验时间点内和实验时间点之间的转变,这是我们用来恢复在咽内胚层发育过程中促进髓质胸腺上皮细胞形成的基因的功能。此外,我们能够根据代谢标记数据估计细胞特异性转录和降解率,并将其应用于肠道类器官系统,以描绘分化轨迹并精确定位调控策略。
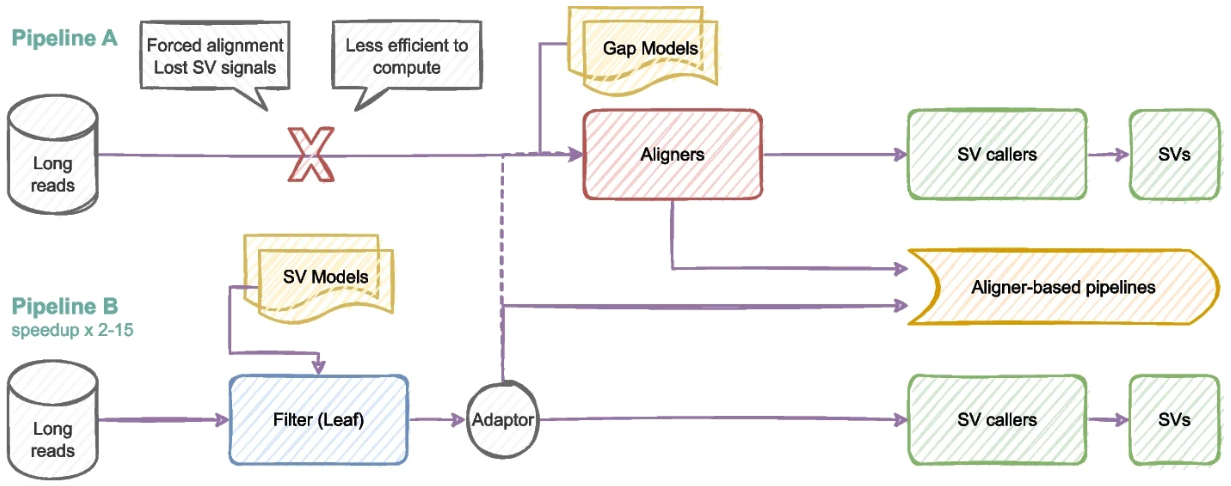
Leaf:用于群体规模长读长 SV 检测的超快过滤器
Leaf: an ultrafast filter for population-scale long-read SV detection – PubMed. Genome Biol
德国柏林自由大学数学与计算机科学系
- 测序技术的进步促进了群体规模的长读长结构变异(SV)检测。可以说,人口规模分析的主要挑战之一是开发有效的计算管道。
- 在这里,我们提出了一种新的基于过滤器的管道Leaf (ALIgNment-free methods for long-read vARiants resolution),用于群体规模的长读长 SV 检测。与传统的基于组装或基于对齐的管道相比,它可以更好地在早期捕获 SV 信号。
- 这项工作的评估表明,基于过滤器的管道有助于更好地解决读内重排。此外,它比传统管道的计算效率更高,因此可以促进人口规模的长读应用。
开发结直肠癌筛查的最佳分层模型并减少多中心人群研究中的种族差异
武汉大学中南医院公共卫生学院流行病学与生物统计学教研室、消化肿瘤科、武汉大学泰康生命与医学中心
- 结直肠肿瘤的早期发现可以通过对高危人群及时干预来减轻结直肠癌(CRC)负担。然而,东亚 (EAS) 人群缺乏有效的风险预测模型来进行个性化 CRC 早期筛查。我们的目标是开发、验证和优化 EAS 人群动态腺瘤-癌序列所有阶段的综合风险预测模型。
- 为了制定精确的风险分层和干预策略,我们开发了三种针对结直肠肿瘤的跨祖先 PRS:(1)使用 148 个先前确定的 CRC 风险位点(PRS148); (2) 通过聚类和阈值处理从大规模荟萃分析数据中选择 SNP (PRS183); (3) PRS-CSx,一种用于全基因组风险预测的贝叶斯方法(PRSGenomewide)。然后,在两个独立的横断面筛查组中评估和验证每个 PRS 的性能,其中包括来自 ZJCRC(浙江结直肠癌组;EAS)的 4600 名晚期结直肠肿瘤患者、4495 名非晚期腺瘤患者和 21,199 名正常个体。 )和 PLCO(前列腺癌、肺癌、结直肠癌和卵巢癌筛查试验;欧洲,EUR)研究。最佳 PRS 进一步与生活方式因素结合起来,对个人风险进行分层,并最终在 PLCO 和英国生物银行前瞻性队列中进行测试,共有 350,013 名参与者。
- 与 EUR 人群相比,三个跨血统 PRS 在 EAS 中的预测性能略有提高。值得注意的是,PRS 有效地促进了动态腺瘤-癌序列所有阶段的彻底风险评估。在这些模型中,PRS183 在 EAS 和 EUR 验证数据集中表现出了最佳的区分能力,特别是对于有结直肠肿瘤风险的个体。使用两个大规模且独立的前瞻性队列,我们进一步证实了 PRS183 对结直肠肿瘤的显着剂量反应效应。与单独使用 PRS 或生活方式因素相比,将 PRS183 与生活方式因素纳入综合策略可提高风险分层和区分准确性。这种全面的风险分层模型显示出解决筛查测试中漏诊问题的潜力(最佳 NPV = 0.93),同时适度减少不必要的筛查(最佳 PPV = 0.32)。
- 我们在基于人群的 CRC 筛查试验中的综合风险分层模型代表了个性化风险评估方面的一个有希望的进步,有助于在 EAS 人群中进行定制的 CRC 筛查。这种方法增强了 PRS 的跨血统可转移性,从而有助于解决健康差异。
(~ ̄▽ ̄)~ 使用机器学习引导的信号富集基于超灵敏等离子体的肿瘤负荷监测
Ultrasensitive plasma-based monitoring of tumor burden using machine-learning-guided signal enrichment – PubMed. Nat Med. full pdf
美国纽约基因组中心
- 在实体瘤肿瘤学中,循环肿瘤 DNA (ctDNA) 有望通过准确评估微小残留病 (MRD) 和治疗反应监测来改变护理方式。为了克服低肿瘤分数(TF)环境中 ctDNA 片段的稀疏性并提高 MRD 敏感性,我们之前通过血浆全基因组测序(WGS)利用全基因组突变整合。
- 我们现在在这里介绍 MRD-EDGE,这是一种机器学习引导的 WGS ctDNA 单核苷酸变异 (SNV) 和拷贝数变异 (CNV) 检测平台,旨在增加信号富集。与之前的 WGS 误差抑制相比,MRD-EDGE SNV 使用深度学习和 ctDNA 特定特征空间,将 WGS 中的 SNV 信噪比富集度提高了约 300 倍。 MRD-EDGECNV 还将通过 WGS 进行超灵敏 CNV 检测所需的非整倍性程度从 1 Gb 降低到 200 Mb,从而大大扩展了其在实体瘤中的适用性。
- 我们利用改进的性能来识别多种癌症类型手术后的 MRD,跟踪肺癌新辅助免疫治疗引起的 TF 变化,并证明癌前结直肠腺瘤中的 ctDNA 脱落。
- 最后,MRD-EDGESNV 中彻底的信噪比丰富使得能够对晚期黑色素瘤和肺癌进行仅血浆(非肿瘤信息)疾病监测,从而为免疫检查点抑制患者提供具有临床信息的 TF 监测。
(~ ̄▽ ̄)~ tidyomics 生态系统:增强组学数据分析
The tidyomics ecosystem: enhancing omic data analyses – PubMed. Nat Methods. full pdf
澳大利亚维沃尔特和伊丽莎·霍尔医学研究所、墨尔本大学医学生物学系
- 组学数据的增长给数据操作、分析和集成带来了不断变化的挑战。为了应对这些挑战,Bioconductor 提供了一个广泛的社区驱动的生物数据分析平台。同时,整洁的 R 编程提供了革命性的数据组织和操作标准。
- 在这里,我们展示 tidyomics 软件生态系统,将 Bioconductor 与 tidy R 范式连接起来。该生态系统旨在简化组学分析、简化学习并鼓励跨学科合作。
- 我们通过分析人类细胞图谱中的 750 万个外周血单核细胞,涵盖六个数据框架和十个分析工具,证明了 tidyomics 的有效性。
Starfysh 整合空间转录组和组织学数据以揭示异质肿瘤免疫中心
Starfysh integrates spatial transcriptomic and histologic data to reveal heterogeneous tumor-immune hubs – PubMed. Nat Biotechnol
- 空间解析的基因表达谱提供了对组织组织和细胞间串扰的深入了解;然而,基于测序的空间转录组学(ST)缺乏单细胞分辨率。目前的 ST 分析方法需要单细胞 RNA 测序数据作为严格解释细胞状态的参考,大多数不使用相关的组织学图像,并且无法推断多个组织之间的共享邻域。
- 在这里,我们展示了 Starfysh,一个使用深度生成模型的计算工具箱,该模型结合了原型分析和任何已知的细胞类型标记,无需单细胞参考即可表征已知或新的组织特异性细胞状态。 Starfysh 使用组织学图像改进了复杂组织中空间动力学的表征,并能够将生态位作为跨组织的空间中心进行比较。
- 对原发性雌激素受体(ER)阳性乳腺癌、三阴性乳腺癌(TNBC)和化生性乳腺癌(MBC)组织进行综合分析,确定了具有患者和疾病特异性细胞类型组成的空间中心,并揭示了代谢重编程塑造侵袭性 MBC 中的免疫抑制中枢。
使用 454,727 个英国生物库全外显子序列表征的跨祖先 CFTR 变异的多样性
- 对囊性纤维化跨膜导电调节因子(CFTR)基因中变异多样性的了解有限,这限制了推进囊性纤维化(CF)分子诊断的努力。这种情况可能导致诊断延迟,随后使患者的健康状况恶化。因此,表征CFTR变异在不同祖源中的谱系对于革新CF的分子诊断至关重要。
- 我们分析了454,727个英国生物银行(UKBB)的全外显子序列,以表征CFTR变异在不同祖源中的多样性。使用PanUKBB分类,参与者被分为六个主要群体:非洲(AFR)、美洲/美洲混血(AMR)、中南亚(CSA)、东亚(EAS)、欧洲(EUR)和中东(MID)。我们分离了特定于祖源的CFTR变异,包括那些引起CF或临床相关的变异。确定了某些引起CF的变异的年龄,并分析了选择压力的影响,对携带临床相关CFTR基因型的参与者进行了表型分析。
- 我们在六个祖源中检测到超过4000个CFTR变异,包括新的特定于祖源的变异。欧洲人拥有最多的独特CFTR变异[n = 2212],而美洲群体拥有最少的独特变异[n = 23]。F508del是除EAS外所有祖源中发现最普遍的引起CF的变异,在EAS中,V520F是最普遍的。EAS的常见变异如3600G > A、V456A和V520分别出现在大约270、215和338代之前,这些变异没有显示出选择压力的证据。16名参与者携带两个引起CF的变异,其中两人被诊断为CF。我们发现154名参与者携带一个引起CF的变异和不同的临床后果(VCC)变异。对携带多个临床相关变异的参与者进行的表型分析显示,与CF及其肺部表型有显著关联[Bonferroni校正后的p < 0.05]。
- 我们利用UKBB数据库全面表征了CFTR变异在不同祖源中的广泛谱系。检测到的超过4000个CFTR变异,包括几种特定于祖源和未表征的CFTR变异,这表明有必要进一步表征它们的功能和临床相关性。总体来说,未被诊断为CF的参与者中出现经典CF表型的情况表明,他们可能会从当前的CFTR调节剂治疗中受益。
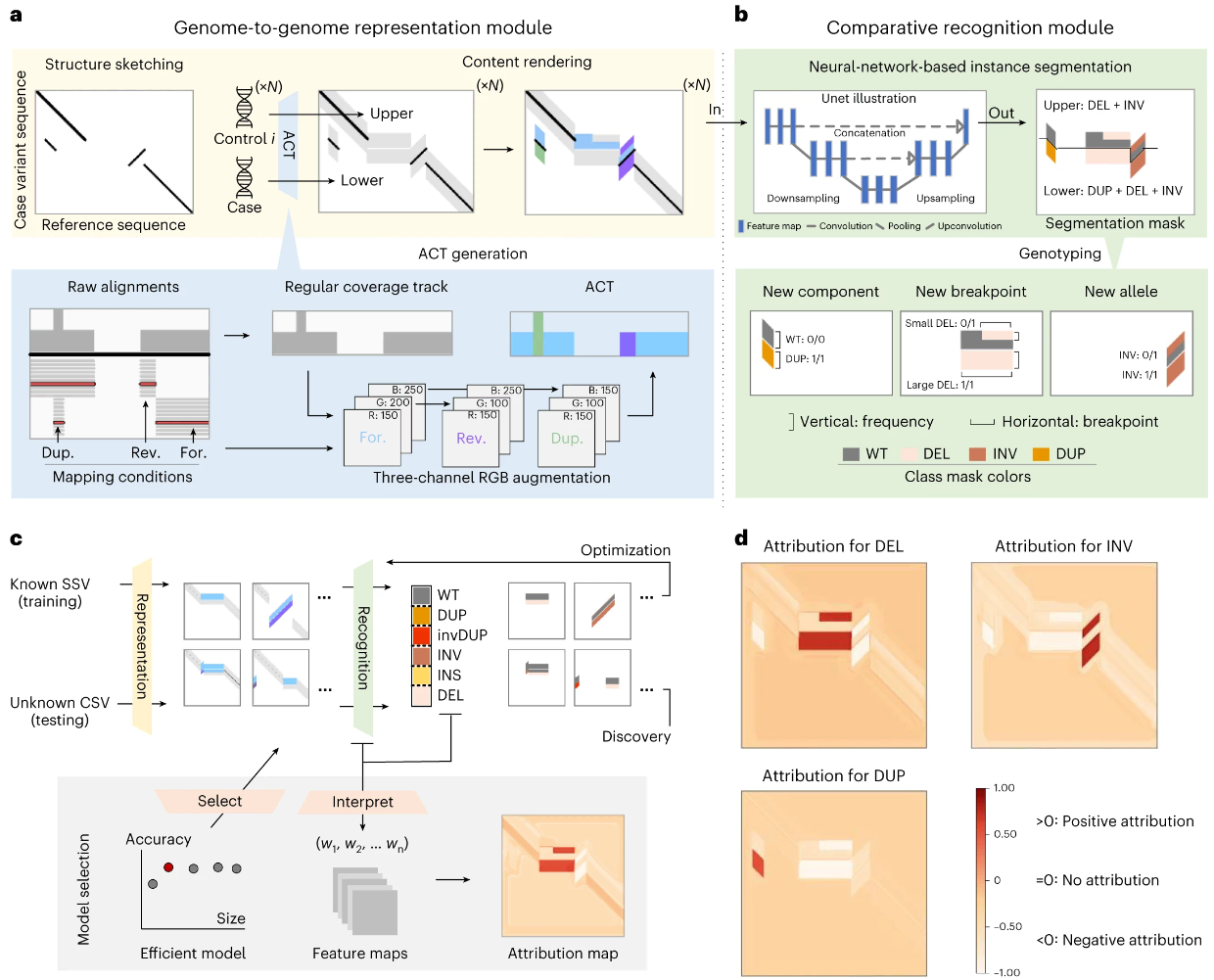
使用 SVision-pro 发现从头和体细胞结构变异
De novo and somatic structural variant discovery with SVision-pro – PubMed. Nat Biotechnol
- 基于长读的从头和体细胞结构变异(SV)发现仍然具有挑战性,需要样本之间的基因组比较。
- 我们开发了 SVision-pro,这是一种基于神经网络的实例分割框架,可以直观地表示基因组到基因组级别的测序差异,并在没有任何推理模型先决条件的情况下发现基因组之间的 SV 比较。
- SVision-pro 优于最先进的方法,特别是复杂 SV 的解析得到了改进,与 SV 合并方法相比,孟德尔错误率低,低频 SV 灵敏度高,误报率降低。
下一代 GWAS:完整的 2D 上位相互作用图检索部分缺失的遗传力并改进表型预测
- 遗传性缺失(missing heritability)的问题需要考虑不同基因座之间的遗传相互作用,称为上位性(epistasis)。目前的 GWAS 统计模型需要数年时间才能评估单个表型的整个组合上位空间。
- 我们提出二代 GWAS (NGG),可在数小时内评估超过 600 亿个单核苷酸多态性组合一级相互作用。
- 我们将 NGG 应用于拟南芥,提供基因分辨率的二维上位图。我们在几种表型上证明,大部分缺失的遗传力可以被检索,它确实存在于上位相互作用中,并且它可以用于改进表型预测。
(~ ̄▽ ̄)~ 评估 GPT-4 在单细胞 RNA-seq 分析中的细胞类型注释
Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis – PubMed. Nat Methods. full html
一个非常取巧的研究 ~ 有空可看看
- 在这里,我们证明大语言模型 GPT-4 可以在单细胞 RNA 测序分析中使用标记基因信息准确注释细胞类型。
- 当对数百种组织和细胞类型进行评估时,GPT-4 生成的细胞类型注释与手动注释表现出很强的一致性。此功能可以大大减少细胞类型注释所需的工作量和专业知识。
- 此外,我们还开发了 R 软件包 GPTCelltype,用于 GPT-4 的自动细胞类型注释。
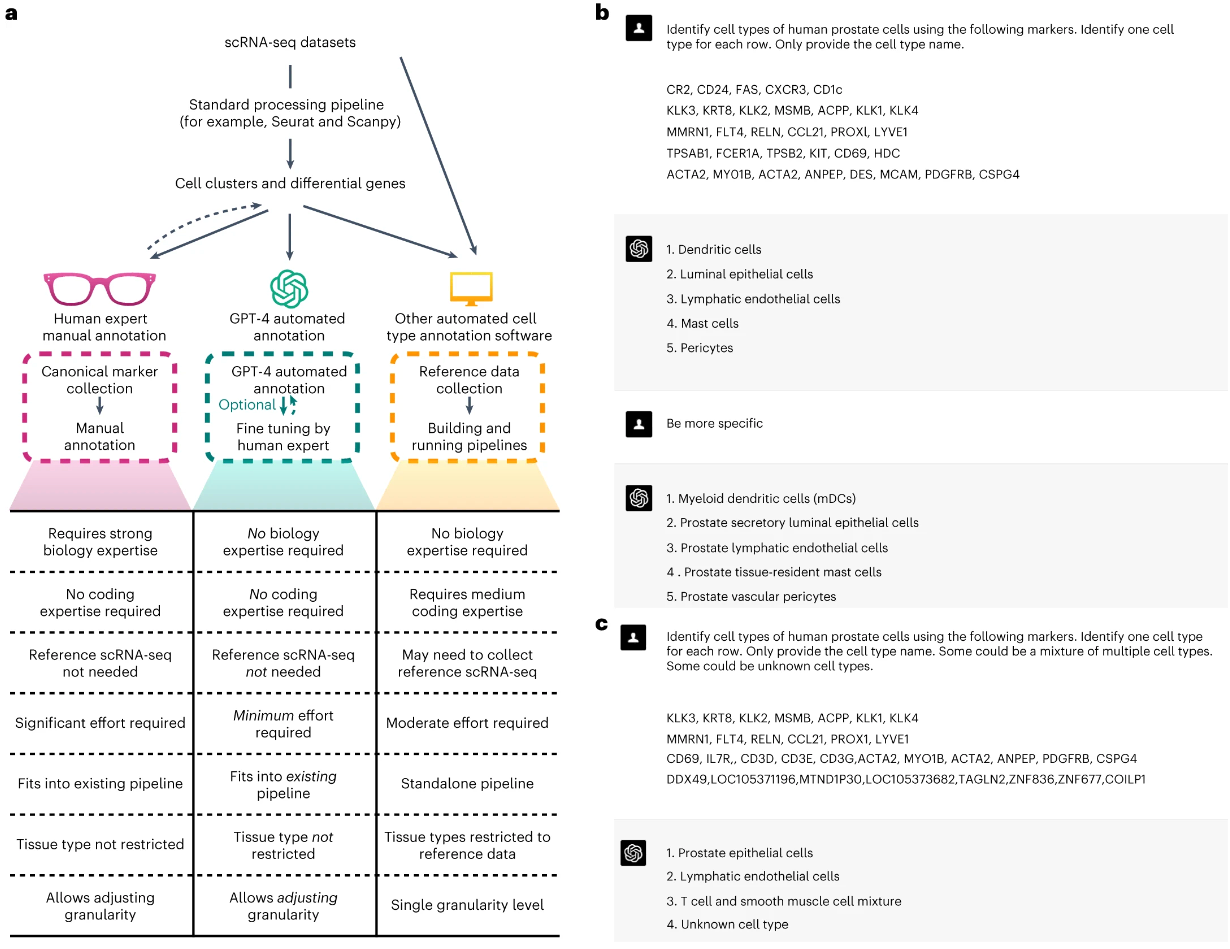
TMO-Net:用于肿瘤学多任务学习的可解释的预训练多组学模型
TMO-Net: an explainable pretrained multi-omics model for multi-task learning in oncology – PubMed. Genome Biol
- 癌症是一种复杂的疾病,由多种尺度的系统性改变组成。
- 在本研究中,我们开发了肿瘤多组学预训练网络(TMO-Net),该网络集成了多组学泛癌数据集进行模型预训练,促进跨组学交互并实现联合表示学习和不完整组学推理。该模型增强了多组学样本表示,并通过不完整的多组学数据集支持各种下游肿瘤学任务。通过采用可解释的学习,我们描述了不同组学特征对临床结果的贡献。
- TMO-Net 模型作为肿瘤学跨模式多组学学习的通用框架,为肿瘤组学特定的基础模型铺平了道路。
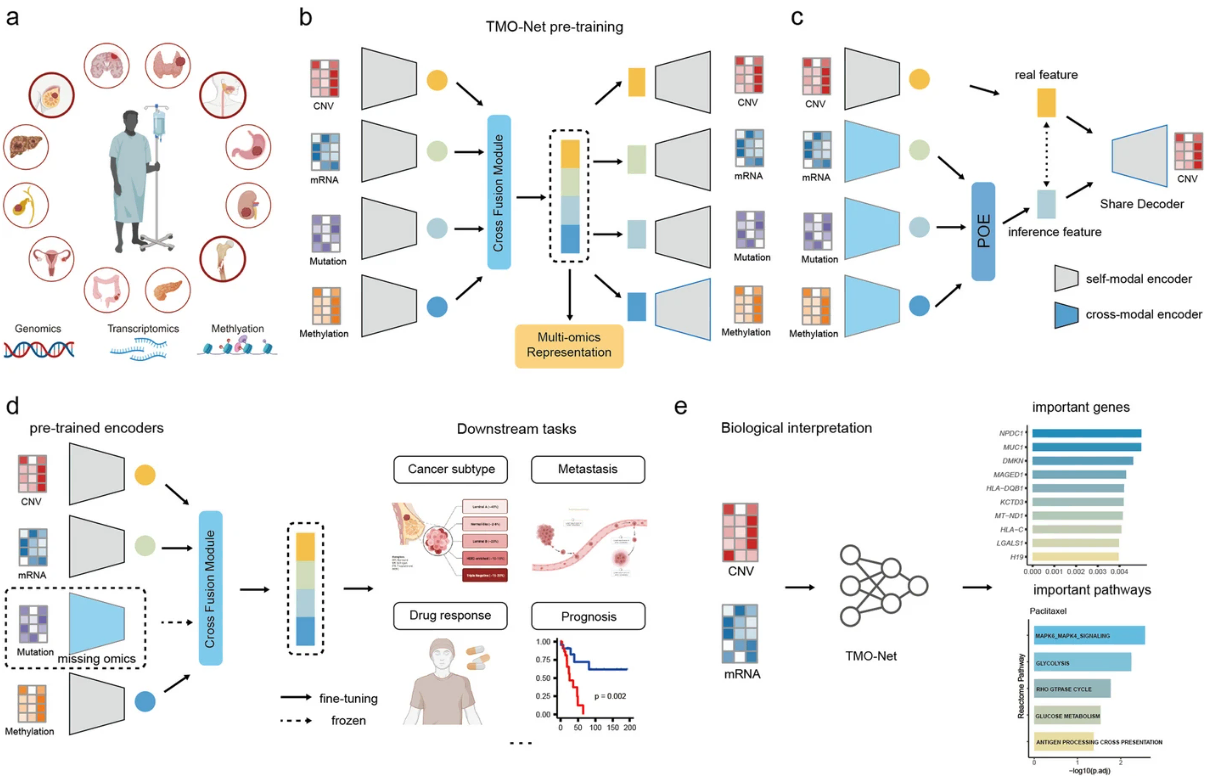
(~ ̄▽ ̄)~ 单细胞转录组学大规模基础模型
Large-scale foundation model on single-cell transcriptomics – PubMed. Nat Methods. full pdf
清华大学自动化系 – 生物信息学部、生物信息学教育部重点实验室
重点关注一下方法学。相关报道:Nat Methods | 1亿个参数,2万个基因!张学工/马剑竹/宋乐开发单细胞转录组学大规模预训练模型
- 大型预训练模型已成为自然语言处理及相关领域取得突破的基础模型。开发破译细胞“语言”和促进生物医学研究的基础模型是有前途的,但也充满挑战。
- 在这里,我们开发了一个大型预训练模型 scFoundation,也称为“xTrimoscFoundationα”,具有覆盖约 20,000 个基因的 1 亿个参数,并针对超过 5000 万个人类单细胞转录组图谱进行了预训练。
- scFoundation 就可训练参数的大小、基因的维度和训练数据量而言是一个大模型,其不对称的类似transformer架构和预训练任务设计能够有效捕获各种细胞类型和状态中基因之间的复杂上下文关系。
- 实验证明了它作为基础模型的优点,在各种单细胞分析任务中实现了最先进的性能,例如基因表达增强、组织药物反应预测、单细胞药物反应分类、单细胞扰动预测、细胞类型注释和基因模块推断。
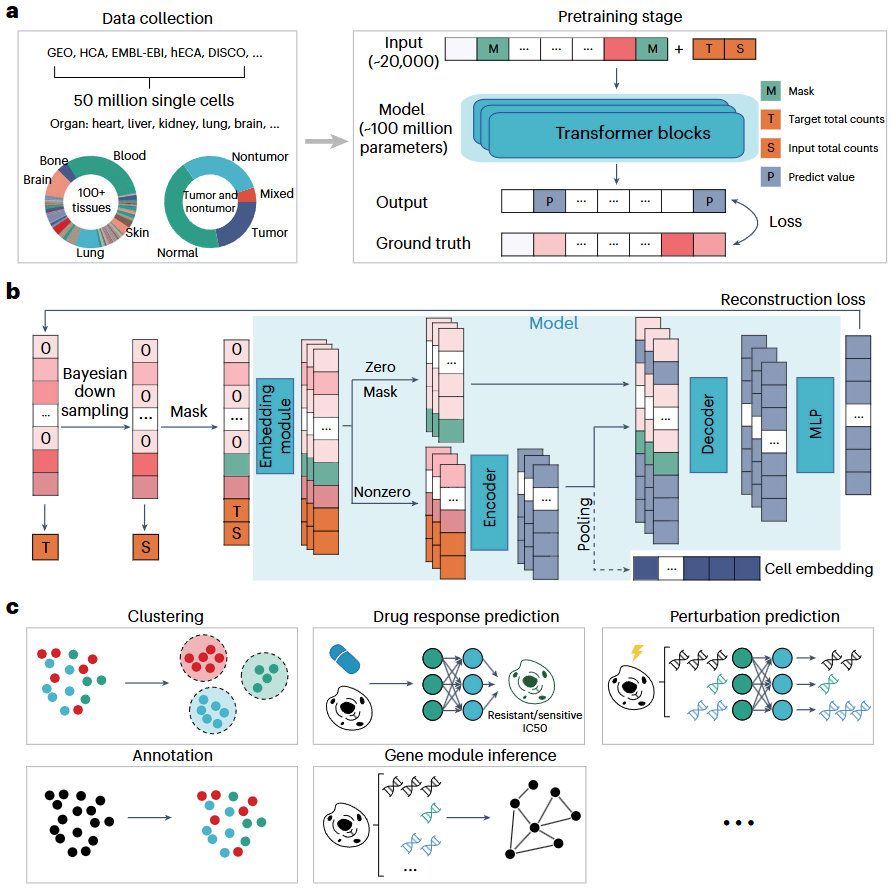
iIMPACT:整合图像和分子概况以进行空间转录组学分析
iIMPACT: integrating image and molecular profiles for spatial transcriptomics analysis – PubMed. Genome Biol
美国德克萨斯大学达拉斯分校数学科学系
- 当前空间转录组数据的聚类分析主要依赖于分子信息,未能充分利用组织学图像中存在的形态特征,导致准确性和可解释性受损。
- 为了克服这些限制,我们开发了一种称为 iIMPACT 的多阶段统计方法。它根据人工智能重建的组织学图像和基因表达测量的空间背景来识别和定义基于组织学的空间域,并检测特定域的差异表达基因。
- 通过多个案例研究,我们证明 iIMPACT 在准确性和可解释性方面优于现有方法,并提供了对空间转录组数据中的细胞空间组织和功能基因景观的见解。
转录因子结合区的 DNA 甲基化模式表征了其功能和进化背景
- DNA甲基化是一种重要的表观遗传修饰,在调节基因组功能中具有多种作用。其水平在整个基因组中具有空间相关性,通常在抑制区域中较高,但在转录因子 (TF) 结合位点和活性调节区域中较低。然而,建立全基因组和 TF 结合位点甲基化模式的机制仍不清楚。
- 在这里,我们使用比较方法来研究哺乳动物中 DNA 甲基化与 TF 结合进化的关联。具体来说,我们通过实验分析了 DNA 甲基化,并将其与已发表的五种哺乳动物(人、猕猴、小鼠、大鼠、狗)肝脏中五种不同 TF(CTCF、CEBPA、HNF4A、ONECUT1、FOXA1)的占用图谱相结合。
- TF 结合位点甲基化程度较低,但它们通常也具有中等甲基化水平。此外,即使核心结合基序中不存在 CpG,结合位点也会受到更广泛结合区域中 CpG 甲基化状态的影响。采用分类和聚类方法,我们提取了 TF 结合区域的 DNA 甲基化水平的独特且物种保守的模式。 CEBPA、HNF4A、ONECUT1 和 FOXA1 具有相同的甲基化模式,而 CTCF 则不同。这些模式表征了 TF 结合区域的替代功能和染色质景观。利用我们的系统发育框架,我们发现 TF 占据的进化丧失导致 DNA 甲基化增加,这表明协调进化。此外,每种甲基化模式都有自己的进化轨迹,反映其基因组背景。
- 我们的表观基因组分析表明 DNA 甲基化在跨物种 TF 结合变化中发挥作用,包括特定的 DNA 甲基化谱表征 TF 结合并与其调控活性、染色质环境和进化轨迹相关。
综述评论类
(~ ̄▽ ̄)~ 机器学习整合方法推进计算免疫学
Machine learning integrative approaches to advance computational immunology – PubMed. Genome Med
德国慕尼黑亥姆霍兹中心-计算生物学研究所Fabiola Curion等
- 免疫学研究传统上依赖蛋白质组学来评估个体免疫细胞,单细胞 RNA 测序已经彻底改变了免疫学研究。计算免疫学家在分析这些数据集方面发挥着至关重要的作用,超越了传统的蛋白质标记识别,涵盖了细胞表型及其功能作用的更详细视图。
- 最近的技术进步允许同时测量单细胞内的多个细胞成分——转录组、蛋白质组、染色质、表观遗传修饰和代谢物,包括组织内的空间环境。这导致了复杂的多尺度数据集的生成,其中可以包括来自相同细胞的多模态测量或配对和不配对模态的混合。现代机器学习 (ML) 技术允许集成多个“组学”数据,而不需要对每种模式进行广泛的独立建模。
- 本综述重点关注应用于免疫学研究的机器学习综合方法的最新进展。我们强调这些方法在创建多尺度数据集合的统一表示方面的重要性,特别是对于单细胞和空间分析技术。最后,我们讨论了这些整体方法的挑战,以及它们如何有助于开发多尺度研究的共同坐标框架,从而加速计算免疫学领域的研究并实现发现。
了解方法的来源和质量对于负责任地重用 FAIR 数据至关重要
Understanding the provenance and quality of methods is essential for responsible reuse of FAIR data. Nature Med
- data findable, accessible, interoperable and reusable
癌症基因组学和组织病理学中的深度学习
Deep learning in cancer genomics and histopathology – PubMed. Genome Med
- 组织病理学和基因组分析是精准肿瘤学的基石,并且常规用于癌症患者。传统上,组织病理学切片由高度训练的病理学家手动审查。另一方面,基因组数据由设计的计算管道进行评估。在这两种应用中,现代人工智能方法,尤其是机器学习(ML)和深度学习(DL)的出现,为从原始数据中提取可操作的见解开辟了一条全新的途径,这可能会增强并可能替代传统评估工作流程的某些方面。
- 在本综述中,我们总结了DL在组织病理学和基因组学中的当前和新兴应用,包括基本诊断任务以及高级预后任务。基于越来越多的证据,我们认为DL可能成为肿瘤学和癌症研究中新型工作流程的基础。
- 然而,我们也指出DL模型可能存在偏见和其他缺陷,医疗和研究领域的用户需要了解这些问题,并提出了解决这些问题的方法。
科普类
种系 CDH1 变异和终生癌症风险
Germline CDH1 Variants and Lifetime Cancer Risk – PubMed. JAMA
Cadherin-1 或上皮钙粘蛋白 (E-cadherin)(不要与 APC/C 激活蛋白 CDH1 混淆)是人类中由 CDH1 基因编码的一种蛋白质。
- 重要性:大约 1% 至 3% 的胃癌和 5% 的小叶乳腺癌是遗传性的。功能丧失 CDH1 基因变异是与遗传性弥漫性胃癌和小叶乳腺癌相关的最常见基因变异。此前,CDH1 基因变异功能丧失的个体终生罹患胃癌的风险估计约为 25% 至 83%,乳腺癌的终生风险估计约为 39% 至 55%。
- 目的:描述 CDH1 变异个体的胃癌和乳腺癌风险估计。设计、背景和参与者:2021 年 1 月至 2022 年 8 月期间对来自北美的 213 个家庭进行了多中心、回顾性队列和建模研究,其中 1 个或多个家庭成员具有 CDH1 致病性或可能致病性 (P/LP) 变异。主要结果和措施:风险比(HR),定义为变异携带者相对于非携带者的风险,对每种癌症类型进行了估计,并用于计算累积风险和直至 80 岁的每十年生命的风险。
- 结果:总共研究了来自 213 个家庭的 7323 名个体,其中 883 名具有 CDH1 P/LP 变异(先证者中位年龄,53 岁 [IQR,42-62];4% 亚洲人;4% 西班牙裔;85% 非西班牙裔)白人;50% 女性)。在携带CDH1 P/LP变异的个体中,女性携带者中胃癌的患病率为13.9% (123/883),乳腺癌的患病率为26.3% (144/547)。 30 岁时晚期胃癌的估计 HR 为 33.5 (95% CI, 9.8-112),70 岁时为 3.5 (95% CI, 0.4-30.3)。
- 男性和女性携带者患晚期胃癌的终生累积风险分别为10.3%(95% CI,6%-23.6%)和6.5%(95% CI,3.8%-15.1%)。基于家族史的胃癌风险估计表明,有 3 名一级亲属受影响的携带者的外显率约为 38%(95% CI,25%-64%)。女性携带者在 30 岁时罹患乳腺癌的 HR 为 5.7(95% CI,2.5-13.2),在 70 岁时为 3.9(95% CI,1.1-13.7)。女性携带者终生患乳腺癌的累积风险为36.8%(95% CI,25.7%-62.9%)。
- 结论和相关性:来自北美的生殖系CDH1 P/LP变异家庭中,胃癌的累积风险为7%至10%,低于先前描述的水平,女性携带者中乳腺癌的累积风险为37% ,这与之前的估计类似。这些发现为当前对具有种系 CDH1 变异的个体的管理提供了信息。
血压的性别特异性遗传结构
Sex-specific genetic architecture of blood pressure – PubMed
- 血压(BP)特征性别差异的遗传和基因组基础仍未得到大规模研究。
- 在这里,我们利用英国生物银行资源对 BP 性状进行了性别分层和组合性别全基因组关联研究,确定了 1,346 个先前报告的和 29 个新的 BP 性状相关位点。
- 在相关基因座中,412 个为女性特异性(Pfemale ≤ 5 × 10-8;Pmale > 5 × 10-8),142 个为男性特异性(Pmale ≤ 5 × 10-8;Pfemale > 5 × 10-8);这些性别特异性位点富含激素相关转录因子,特别是雌激素受体1。对基因与性别相互作用和性二态效应的分析确定了四个基因组区域,显示了女性特异性与舒张压或脉压的关联,包括染色体 13q34-COL4A1/COL4A2 位点。
- 值得注意的是,女性特有的脉压相关位点在动脉组织中表现出丰富的乙酰化组蛋白 H3 Lys27 修饰,并且与女性特有的纤维肌性发育不良(一种女性偏向的血管疾病)相关。共定位信号包括 Chr13q34:COL4A1/COL4A2、Chr9p21:CDKN2B-AS1 和 Chr4q32.1:MAP9 区域。血压特征的性别特异性和性别偏倚多基因关联与多种心血管特征相关。
- 这些发现表明血压对心血管疾病具有潜在的临床意义和性别特异性多效性。
三重激素受体激动剂瑞他鲁肽治疗代谢功能障碍相关的脂肪肝病:一项随机 2a 期试验
- Retatrutide 是葡萄糖依赖性促胰岛素多肽、胰高血糖素样肽 1 和胰高血糖素受体的新型三重激动剂。一项为期 48 周的 2 期肥胖研究表明,瑞他鲁肽 8 毫克和 12 毫克的体重分别减轻了 22.8% 和 24.2%。该子研究的主要目的是评估患有代谢功能障碍相关脂肪肝病且 LF ≥10% 的研究参与者在 24 周时肝脏脂肪 (LF) 相对于基线的平均相对变化。
- 在这项随机、双盲、安慰剂对照试验中,参与者 (n = 98) 被随机分配接受每周一次皮下注射瑞他鲁肽(1、4、8 或 12 毫克剂量)或安慰剂,为期 48 周。 24 周时 LF 相对于基线的平均相对变化为 -42.9% (1 mg)、-57.0% (4 mg)、-81.4% (8 mg)、-82.4% (12 mg) 和 +0.3%(安慰剂) (与安慰剂相比,所有 P < 0.001)。 24 周时,27%(1 毫克)、52%(4 毫克)、79%(8 毫克)、86%(12 毫克)和 0%(安慰剂)的参与者达到正常 LF(<5%)。
- LF 的减少与体重、腹部脂肪和代谢指标的变化显着相关,而代谢指标与胰岛素敏感性和脂质代谢的改善相关。 ClinicalTrials.gov 注册号为 NCT04881760。
与中国65岁以上成年人热浪死亡相关的危险因素
Risk factors associated with heatwave mortality in Chinese adults over 65 years – PubMed. Nat Med
- 由于生理因素和合并症,老龄化人口很容易遭受与高温相关的死亡。然而,对老龄化人口中个体脆弱性的理解并不完整。
- 在中国健康长寿纵向调查中,我们对 2008 年至 2018 年随访期间的 13,527 名参与者(中位年龄 = 89 岁)和 3,249 名夏季死亡率进行了每日热浪暴露评估。
- 根据相对温度,热浪日期间的死亡风险大约增加了一倍(风险比 (HR) 范围 = 1.78-1.98)。我们发现,对于行动能力下降(HR范围= 2.32-3.20)、日常生活活动依赖(HR范围= 2.22-3.27)、认知障碍(HR = 2.22)和社会孤立的个体来说,热浪死亡风险增加在遇到困难时没有人寻求帮助(HR 范围 = 2.14-10.21)。与目前的认识相反,考虑到个人功能下降后,年龄较大并不能预测热浪死亡风险;没有发现性别之间的统计差异。
- 除了年龄作为危险因素之外,我们的研究结果强调,功能老化是增强热浪抵御能力的一个潜在因素。评估功能衰退和实施护理策略对于有针对性地预防热浪期间的死亡至关重要。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍
