本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概述
此文内容目前处于BETA版本
Coordinated transcriptional and catabolic programs support iron dependent adaptation to RAS-MAPK pathway inhibition in pancreatic cancer. Cancer Discov.
加利福尼亚大学旧金山医学院Rushika M Perera团队
全文资源:full.pdf.local; full.pdf.remote
ferritinophag 铁自噬;
技术
Inhibitors/drugs
- BafA1:巴佛洛霉素A1,大环内酯类抗生素,是一种知名的自噬晚期阶段抑制剂,可阻断自噬体与溶酶体的融合,并抑制培养细胞溶酶体中的酸化和蛋白质降解。在本文中,BafA1作为是特异、可逆的V-ATPase抑制剂用于诱导急性溶酶体抑制。由于BafA1在体内试验中的毒性较大,FDA目前只批准chloroquine (CQ)作为临床实验中的自噬抑制剂。
- Trametinib:在本文中,Trametinib一般统称为
MEKi。曲美替尼是吡啶并嘧啶(作为其二甲基亚砜加成化合物)药物,一种口服生物可利用的丝裂原活化蛋白激酶激酶(MAP2K;MAPK/ERK 激酶;MEK)1 和 2 抑制剂,具有潜在的抗肿瘤活性。口服后,曲美替尼特异性结合并抑制 MEK 1 和 2,从而抑制各种癌症中生长因子介导的细胞信号传导和细胞增殖。 MEK 1 和 2,双特异性丝氨酸/苏氨酸和酪氨酸激酶,通常在各种癌细胞类型中上调,在调节细胞生长的 RAS/RAF/MEK/ERK 信号通路的激活中起关键作用。
Experiments
- In-gel activity assay for Complex I activity in purified mitochondria from KP4 cells
- Blue native PAGE analysis of purified mitochondrial fractions
- Oxygen consumption rate of individual mitochondrial respiratory complex (complexes I-III and II-III)
Molecules
- LAMP2: 即Lysosomal Associated Membrane Protein 2,溶酶体相关膜蛋白2。LAMP2在伴侣介导的自噬中发挥重要作用,该过程介导蛋白质的溶酶体降解以响应各种压力,并作为具有长生物半衰期的蛋白质正常周转的一部分。通过结合目标蛋白(例如 GAPDH 和 MLLT11)并诱导目标蛋白在溶酶体降解来发挥作用。在响应饥饿的溶酶体蛋白降解中发挥作用(通过相似性)。自噬过程中自噬体与溶酶体融合所必需的 。缺乏 LAMP2 的细胞表达正常水平的 VAMP8,但无法在自噬体上积累 STX17,这是自噬体和溶酶体之间缺乏融合最可能的解释。自噬体内容物正常降解所必需的。需要通过其在溶酶体蛋白降解中的功能有效地介导 MHCII 介导的外源抗原呈递;由内体/溶酶体区室中的蛋白酶产生的抗原肽被新生的 MHCII 亚基捕获。不需要有效的 MHCII 介导的内源性抗原呈递 。
- NCOA4:Ferritin is degraded by autophagy-dependent capture via the adaptor NCOA4, a process referred to as ferritinophagy (36, 37). 本文中,NCOA4是铁自噬受体,通过阻断NCOA4达到抑制铁自噬的作用。
预备知识
- KRAS-MAPK通路在胰腺导管癌中广泛激活,但KRAS-MAPK抑制仍面临耐药问题。
- 近期有研究表明KRAS/MEK/ERK抑制可诱导适应性的自噬反应增强从而诱导耐药,但具体的机制仍不明确。
- 研究者前期研究表明主转录因子中的小眼转录因子E(MiT/TFE)家族的MITF、TFE3、TFEB的持续激活可诱导高水平的自噬和溶酶体活性,它们均属于MYC超家族,在结构上有某些相似性。抑制MiT/TFE可降低溶酶体和自噬活性,并显著抑制肿瘤生长。
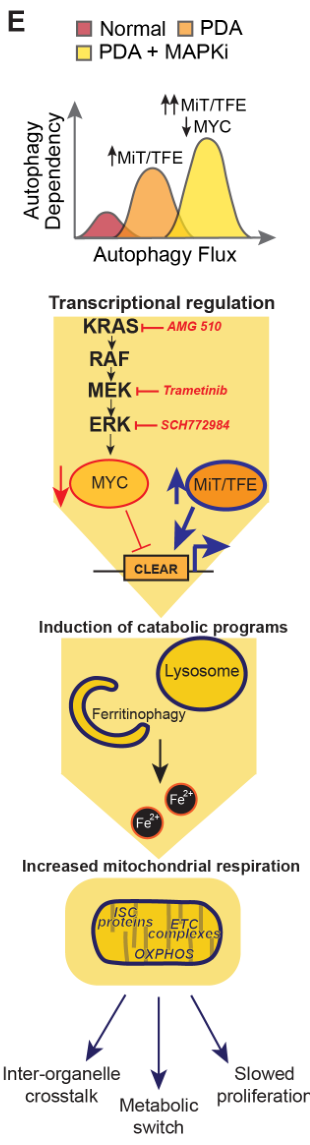
- 本研究主要发现了c-MYC通过MiT/TFE蛋白调控PDA的自噬和溶酶体相关基因。KRAS-MAPK通路受抑制后,c-MYC活性下调,导致MiT/TFE介导的铁自噬(自噬类型之一)和溶酶体信号上调,从而介导PDA对KRAS-MAPK抑制剂耐受。
结果
MEKi诱导自噬和溶酶体基因转录上调的过程依赖TFEB
这一段基本上是在前人研究的基础上,进一步确定“MEKi——自噬”和“MEKi——MiT/TFE——自噬/溶酶体相关基因”的基本调控框架。
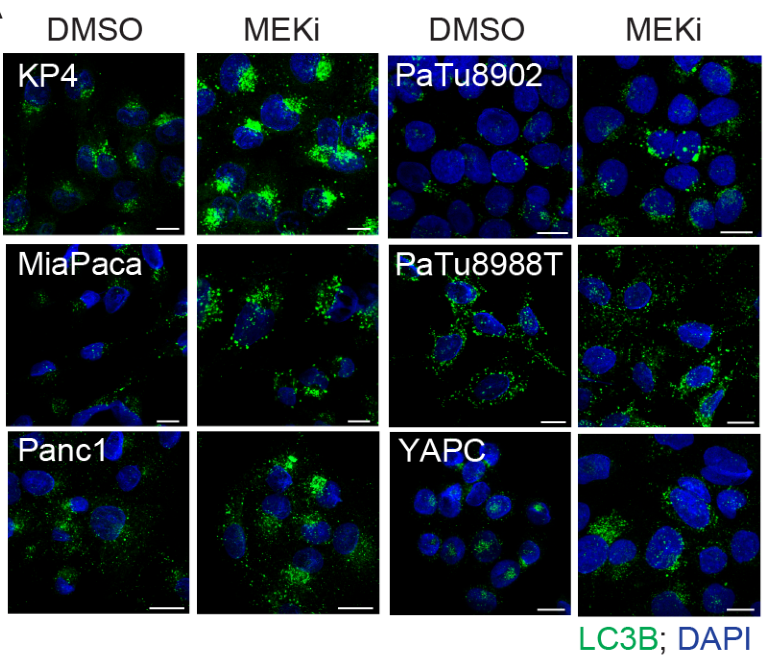
MEKi抑制可促进自噬体蛋白LC3B表达上调和自噬流增加:
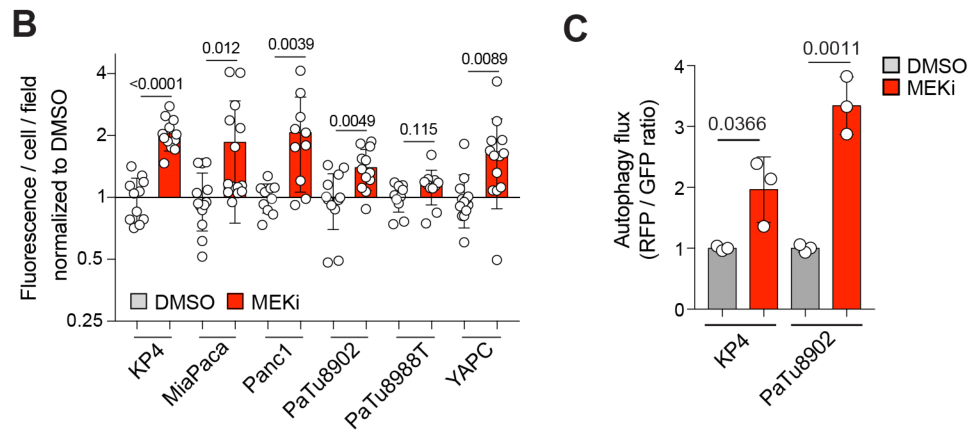
MEKi促进Lysosome相关基因的表达:
MEKi促进自噬-溶酶体相关蛋白LAMP2(自噬体与溶酶体融合的关键蛋白之一)的表达:
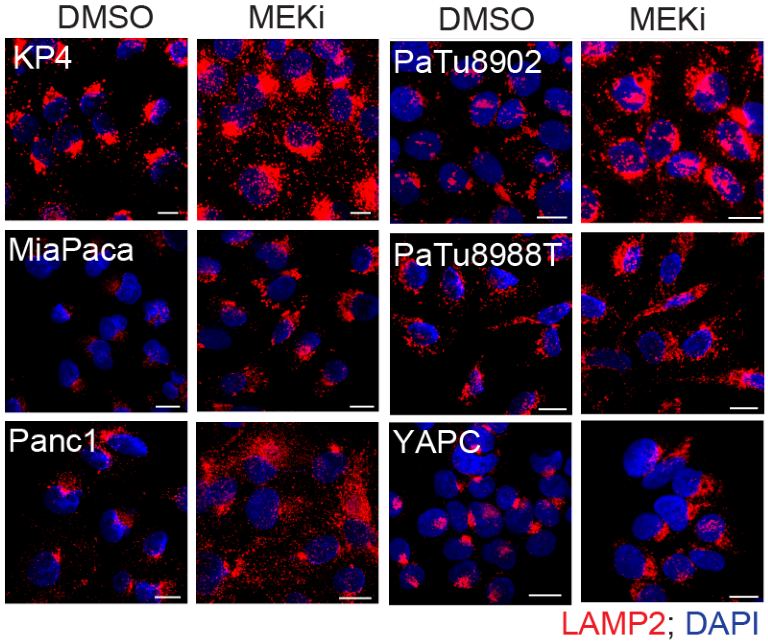
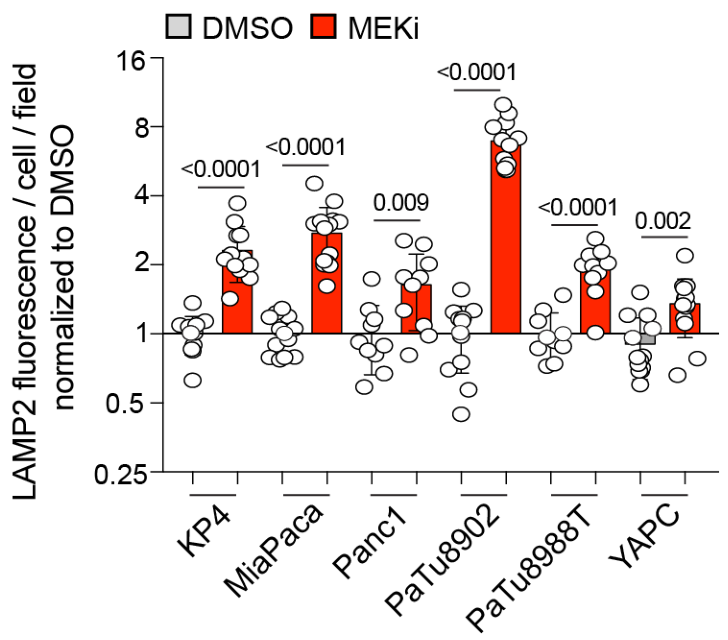
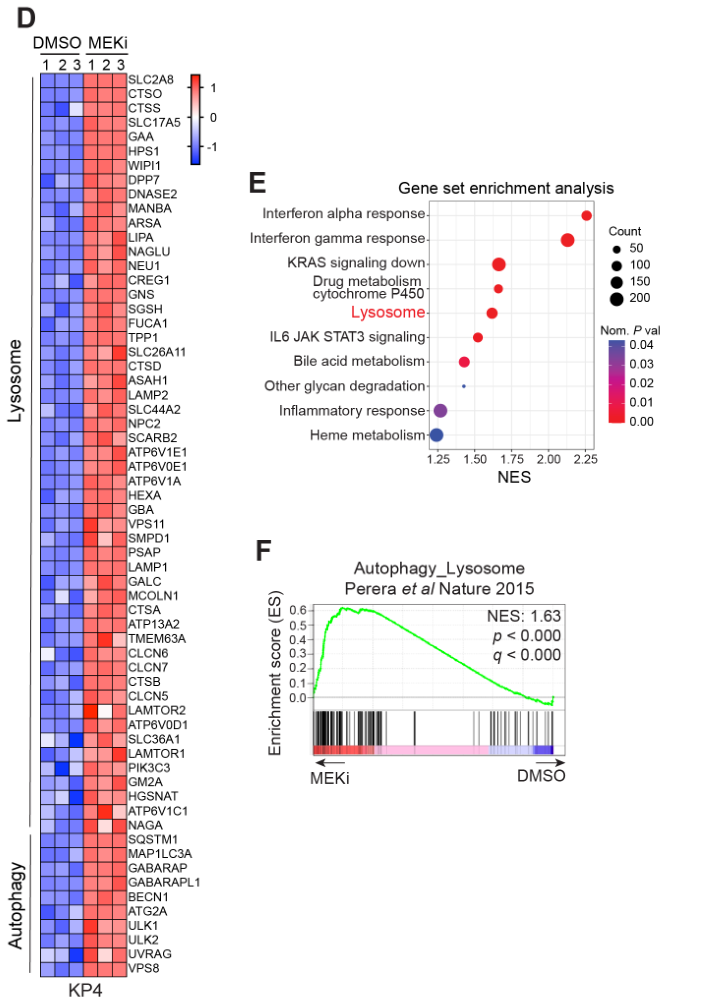
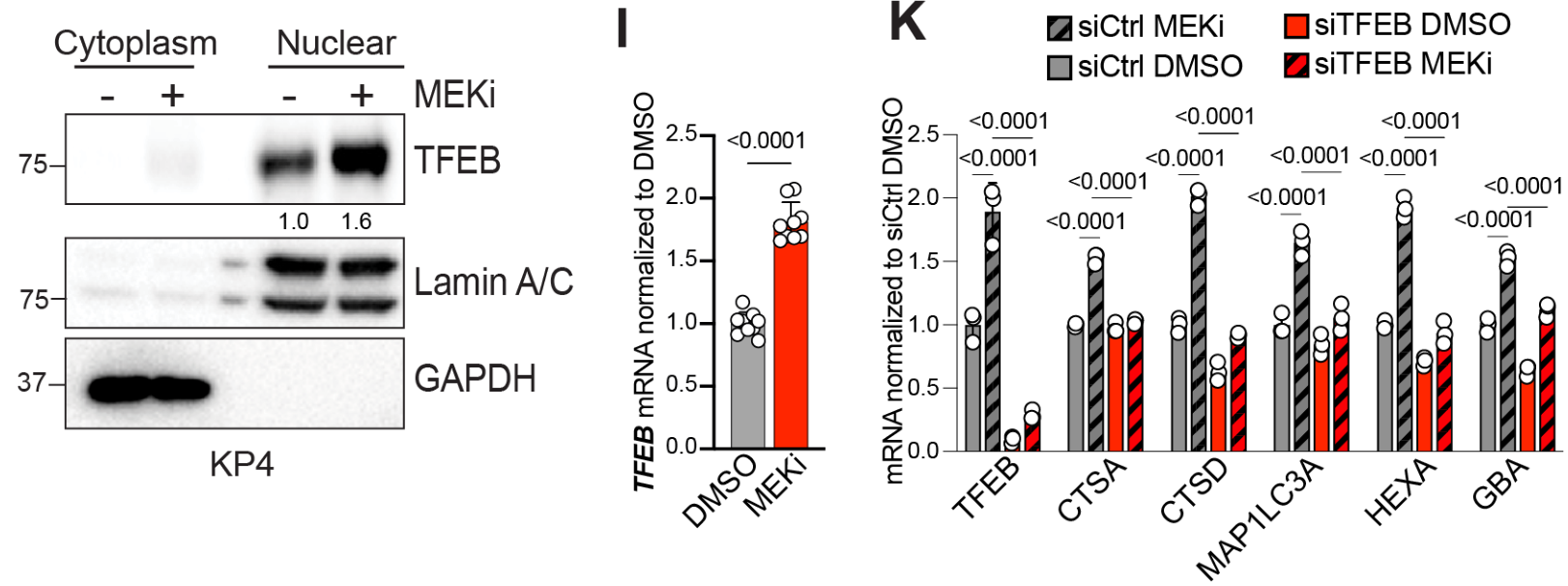
Western Blot和RT-qPCR实验证明MEKi促进自噬和溶酶体相关基因表达,并且此过程依赖TFEB:
由于目标蛋白是核蛋白,Western中检测了核/质分离的样本
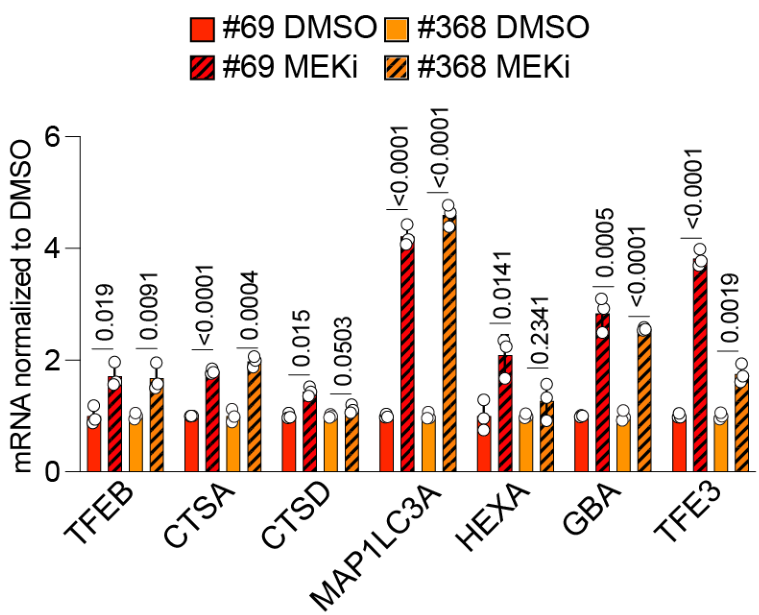
MYC下调促进溶酶体基因表达上调
基本上确立“MEKi——MYC——MYC靶基因”的调控框架
MEKi可抑制MYC下游靶基因的表达:
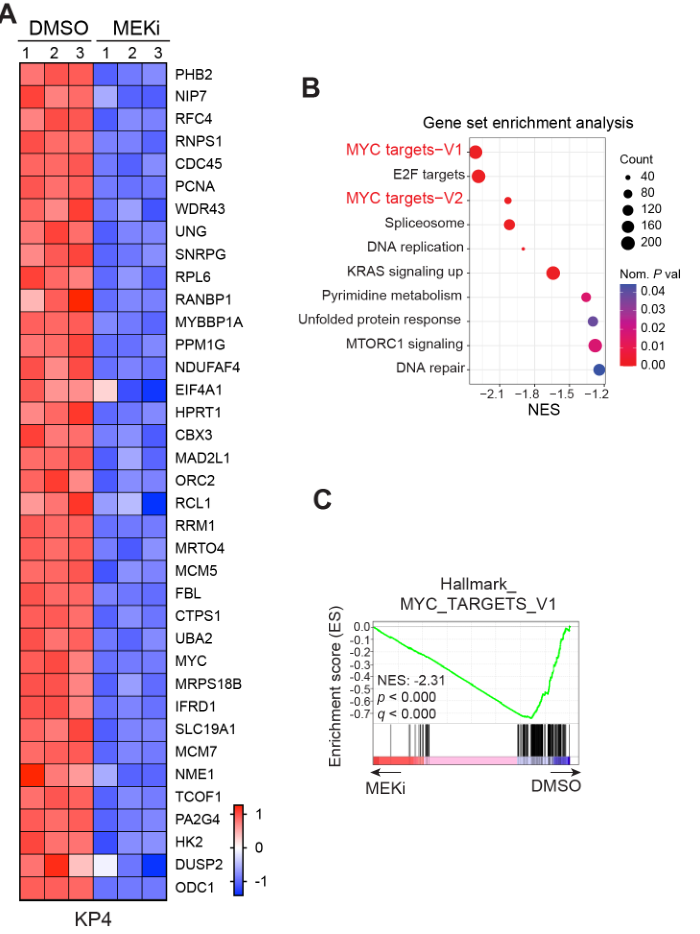
多种实验证实MEKi抑制MYC表达:
观察cMYC-mRNA/protein水平;观察核/质的分布
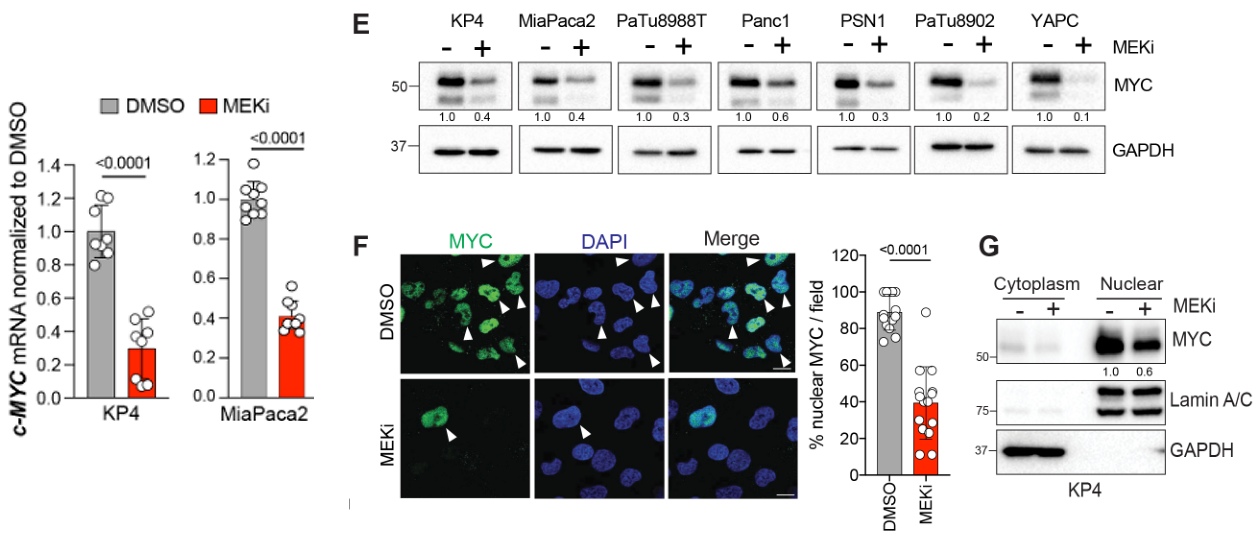
类器官培养和刺激实验也表明MEKi抑制MYC表达:
在3d培养组织中进一步验证
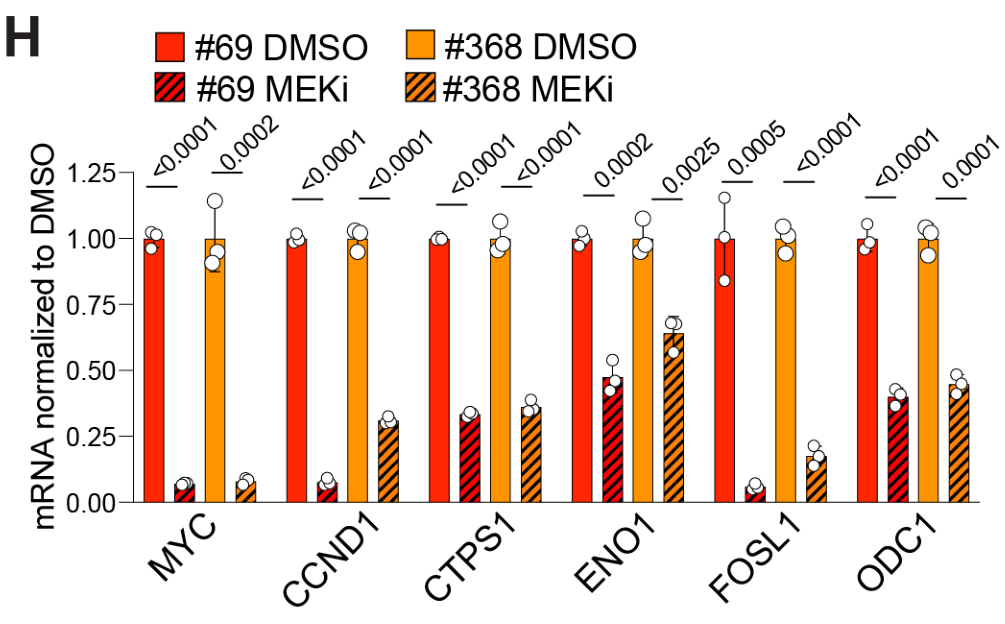
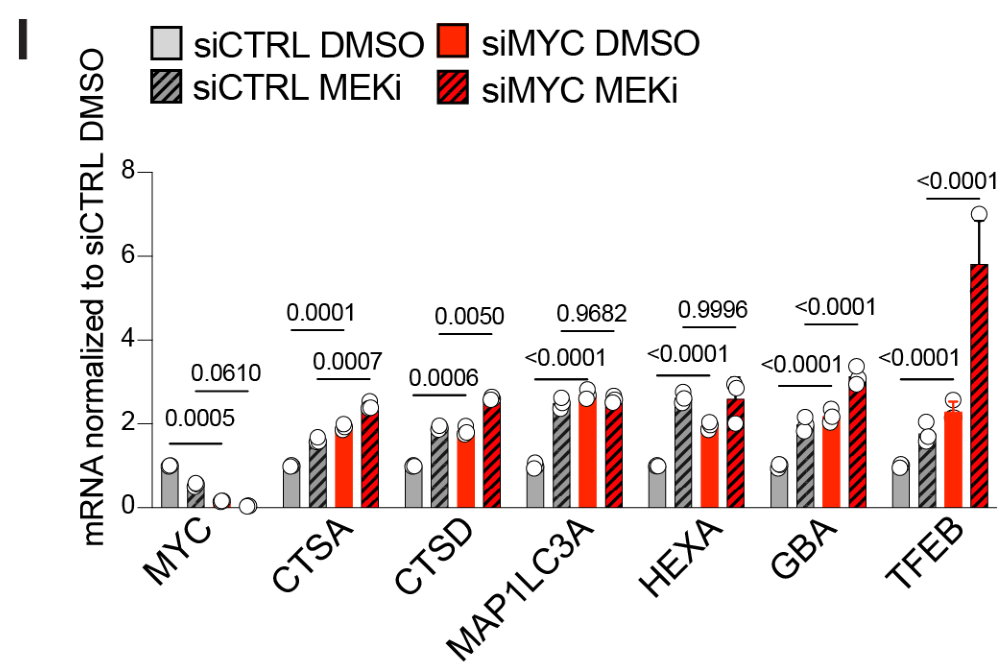
MYC knockdown后,溶酶体基因表达上调;MYC knockdown的情况下,MEKi对溶酶体基因表达上调的幅度变小,这表明MEKi上调溶酶体基因的过程较大程度上依赖MYC:
MEKi诱导溶酶体基因启动子处转录因子的占位变化
本部分主要阐明MYC与MiT/TFE对转录因子的调控具有“此消彼长”的关系。MYC信号弱了,MiT/TFE(主要是基于TFE3的实验)就跟上。
通过通路富集分析发现受转录因子TFE3和MYC调控的下游基因有很多重叠之处:
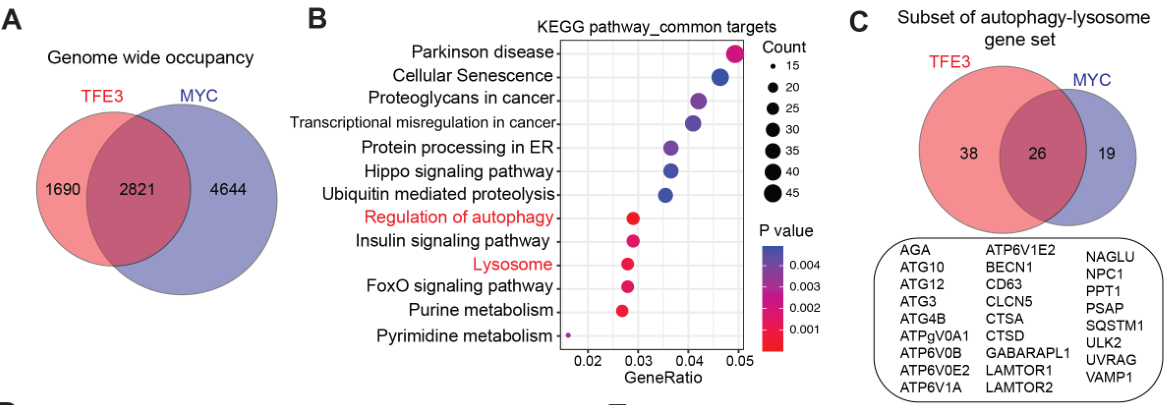
通过ChIP-seq进一步表明MEKi可使MYC与基因TSS的结合减弱,但不影响 TFE3与基因TSS的结合(以ATG3/CTSD/CTSA):
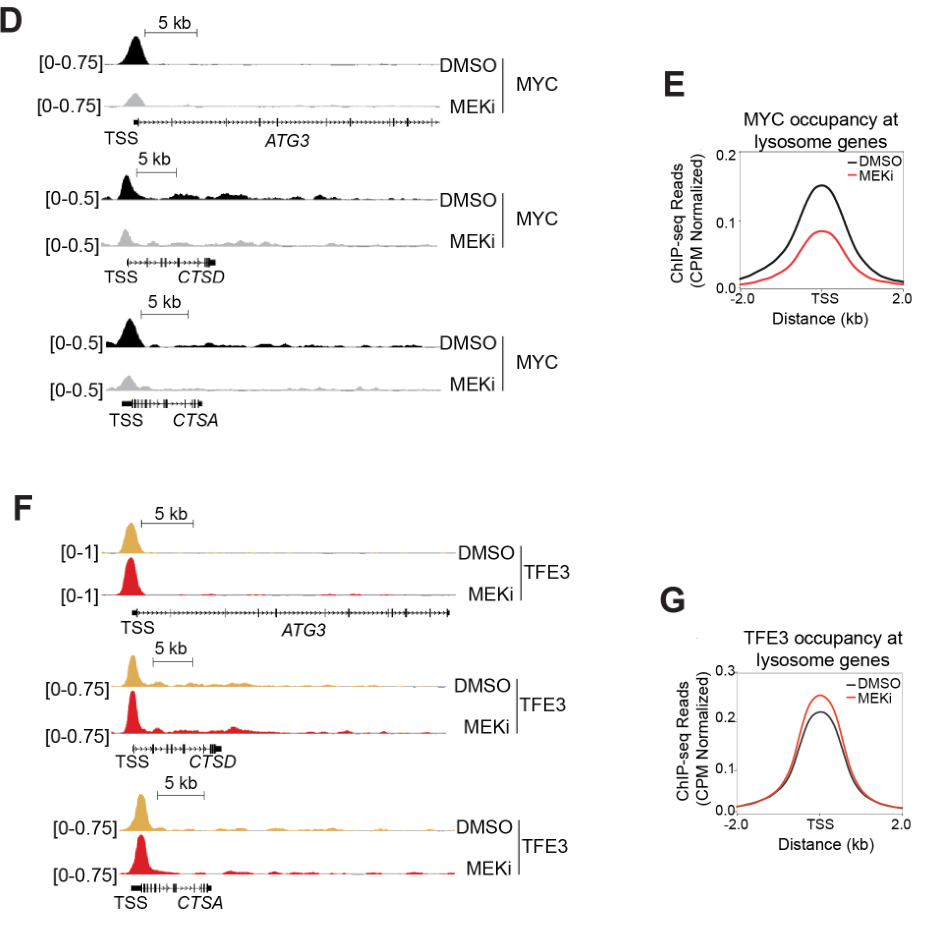
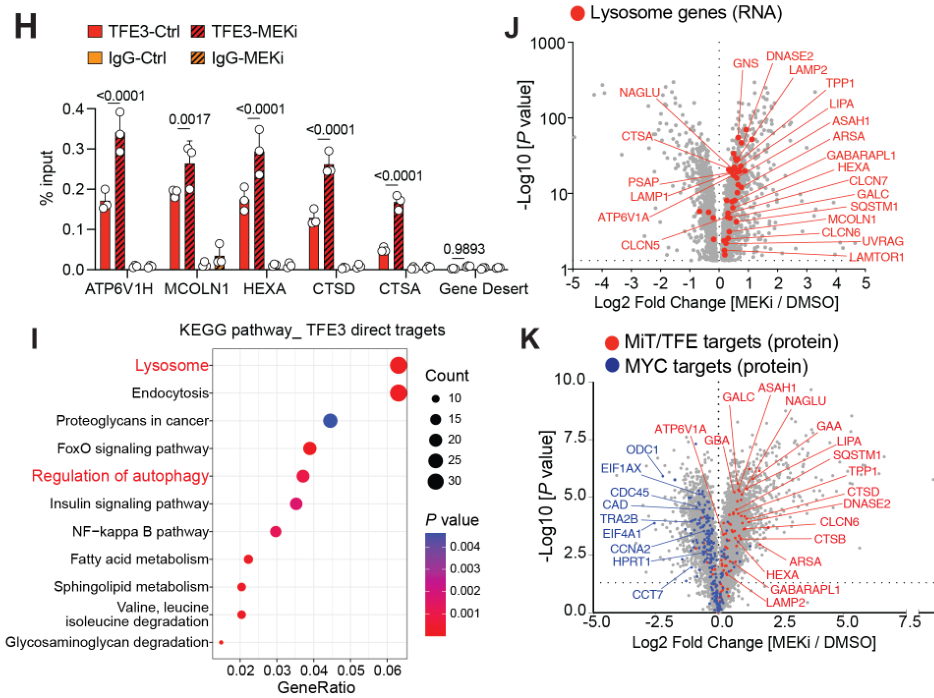
Chiq-Seq+PCR实验表明MEKi处理后TFE3与靶基因TSS结合程度更高(H); 通路富集分析表明MEKi处理后TFE3靶基因表达上调的部分显著富集于溶酶体和自噬相关通路;蛋白组学数据表明MEKi可抑制MYC靶基因的表达并上调MiT/TFE靶基因的表达。
J图怎么感觉与前面的内容有点重复?
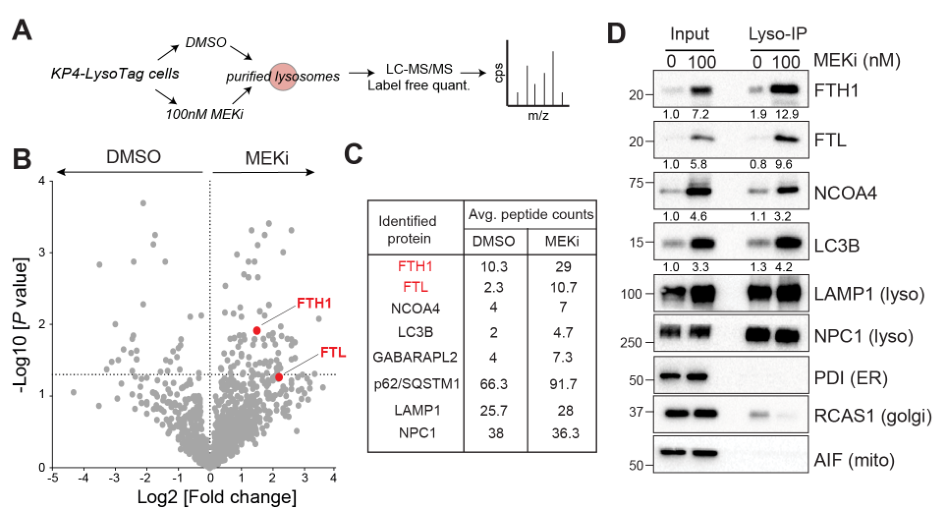
MEKi诱导了铁自噬
本部分主要是明确“MEKi——溶酶体/铁自噬信号上调”的研究框架
提纯溶酶体后,发现MEKi可诱导溶酶体蛋白FTH1和FTL表达上调;通过co-IP实验表明MEKi促进了FTH1/FTL与溶酶体结合:
更近一步地,通过免疫荧光观察到MEKi促进溶酶体相关膜蛋白2(LAMP2)与FTL结合;溶酶体抑制剂(BafA1)可增强溶酶体与FTL的结合?
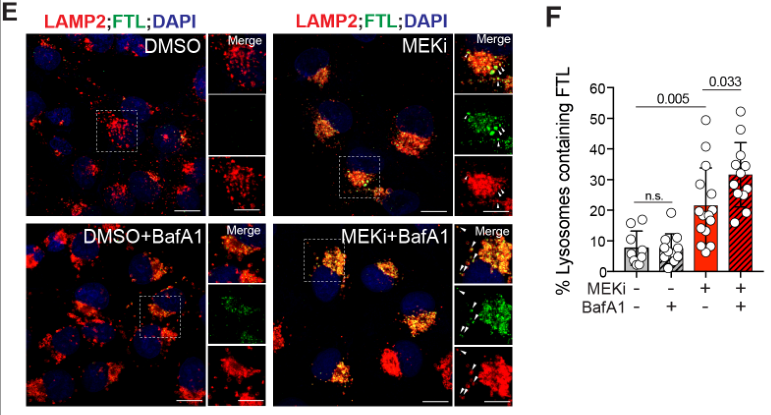
erroOrange iron dye染色表明MEKi促进了铁积聚,但溶酶体抑制剂可抑制这一效应,说明MEKi诱导铁自噬的过程依赖溶酶体活性:
FAS、DFO分别充当阳性、阴性对照
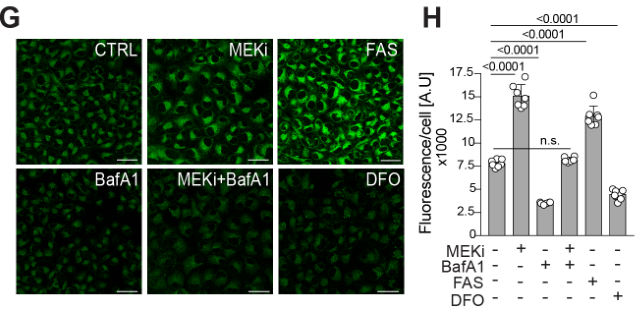
体内外实验证明MEKi可促进FTH1与FTL的总体表达水平:
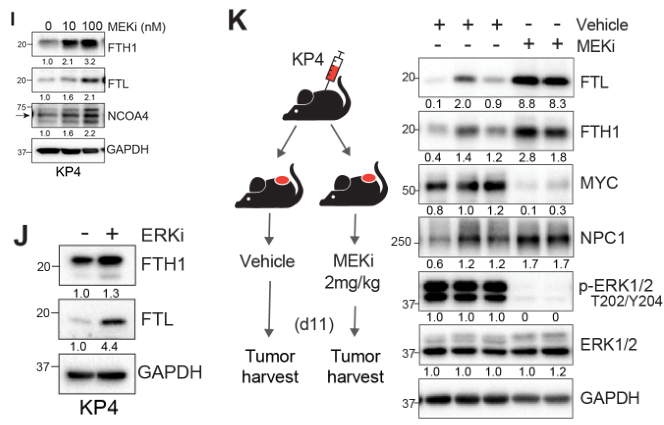
实验表明MEKi降低膜转铁受体Transferrin receptor (TfR1) 含量和Ferroportin 铁输出蛋白 (FPN)的含量,表明MEKi抑制铁原子的外排:
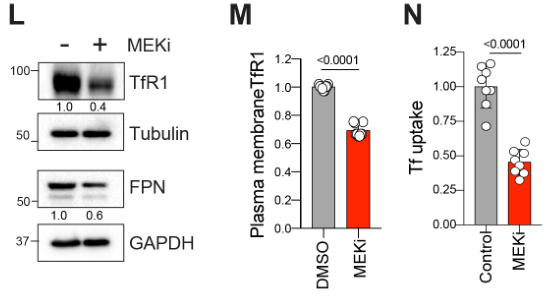
MEKi增强了线粒体铁硫簇蛋白水平、复合物活性和呼吸作用
背景:细胞质不稳定铁池 (labile iron pool, LIP) 里面的氧化还原活性铁用于合成血红素和 ISC 。 ISC 是线粒体电子传递链 (ETC) 的复合体 I、II 和 III 中几种蛋白质的必需辅助因子,而复合体 II、III 和 IV 中的其他蛋白质含有血红素。所以,此部分也是自然而然想到的,主要是为了深化机制研究。
本部分主要是为了证明MEKi对线粒体活性的影响。
全细胞蛋白组学数据显示MEKi诱导多个线粒体铁硫簇蛋白(Iron-sulfur cluster, ISC)表达上调,随后的全细胞和线粒体提纯物的Western blot分析也进一步佐证:
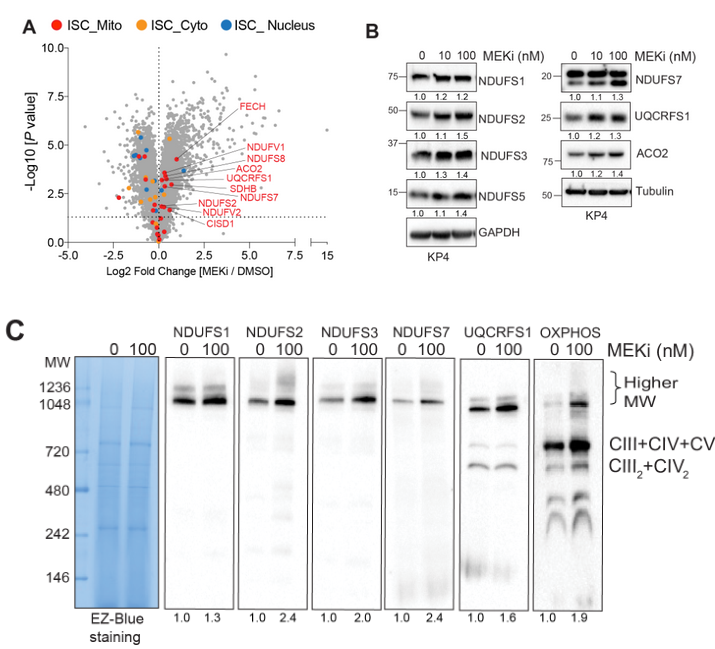
In-gel activity assay表明MEKi促进线粒体呼吸复合物I的活性和ISC蛋白Aconitase 2的活性:
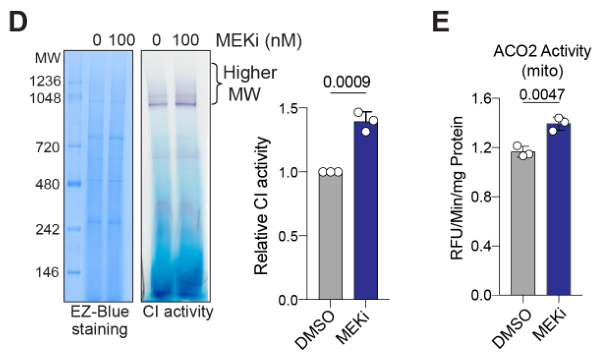
MEKi促进KP4细胞线粒体呼吸复合物I-III/II-III的耗氧率(Oxygen consumption rate, OCR):
Oxygen consumption rate of mitochondrial respiratory complex using seahorse in KP4 cells treated with DMSO or MEKi.
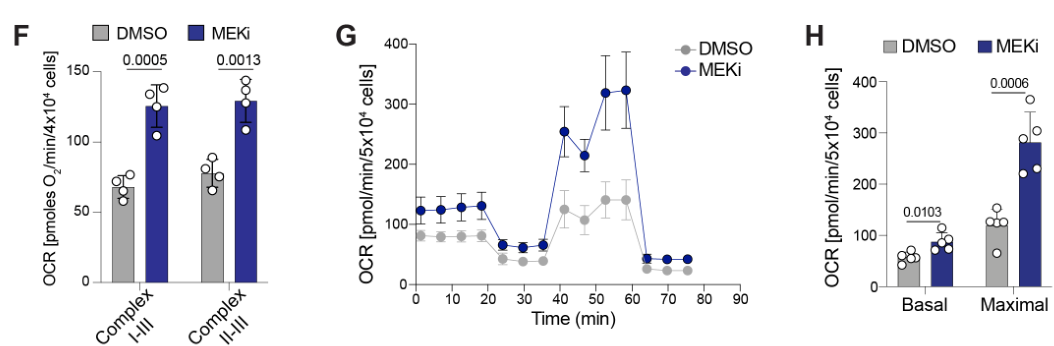
阻断铁自噬或溶酶体会损害线粒体活性和细胞活力
NCOA4 nuclear receptor coactivator 4; 以配体依赖性方式与雄激素受体相互作用以增强其转录活性; 在本文中,NCOA4是铁自噬受体;
exogenous iron, FAC, 外源性铁; Chloroquine, 氯喹, CQ; iron chelator, DFO铁清除剂; FAC诱导铁自噬,DFO抑制铁自噬,两者分别是阳性/阴性对照。
除了表明线粒体活性、ISC上调等MEKi相关效应依赖铁自噬外,还初步展示MEKi+铁自噬抑制的联合治疗有协同效应
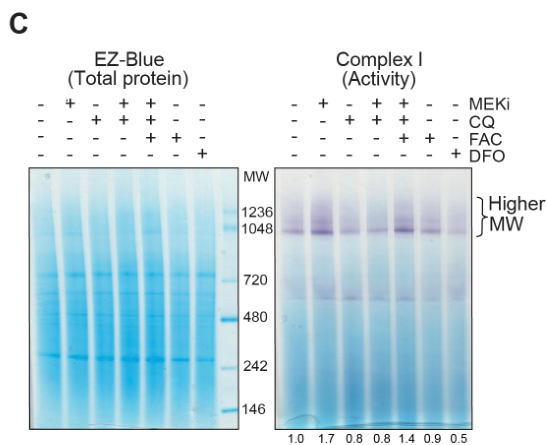
实验表明MEKi介导的ISC上调依赖铁自噬(通过干扰NCOA4控制铁自噬);类似的,自噬抑制剂氯喹也可取消MEKi介导的ISC上调:
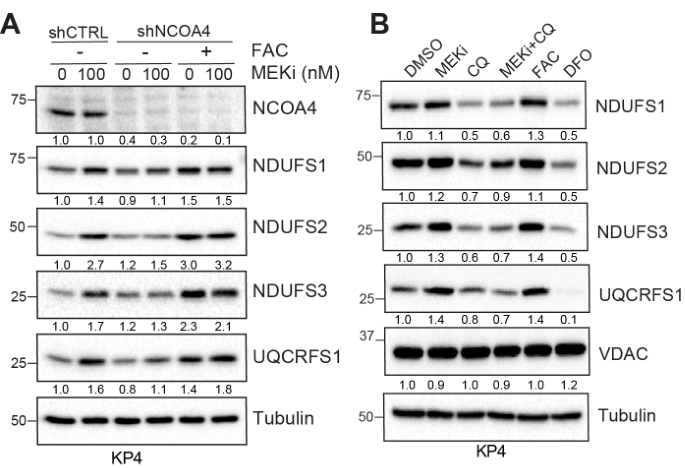
同时,氯喹也可抑制MEKi介导的复合物I活性增强,并且FAC使用可以回复氯喹介导的复合物I活性抑制效应:
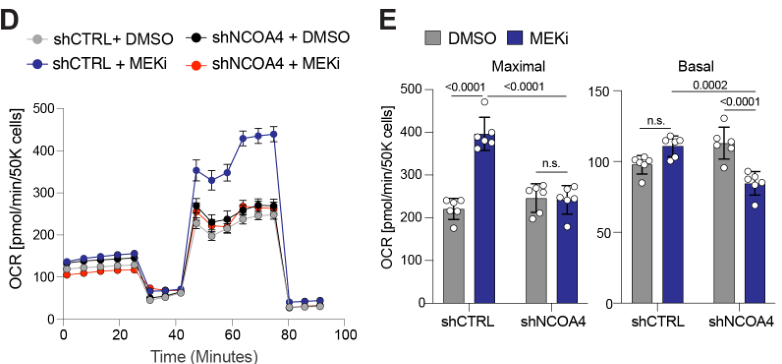
NCOA4 knockdown的情况下,MEKi无法诱导KP4细胞OCR增加,甚至基础OCR还有所下降,这表明MEKi诱导KP4细胞OCR增加的效应依赖铁自噬:
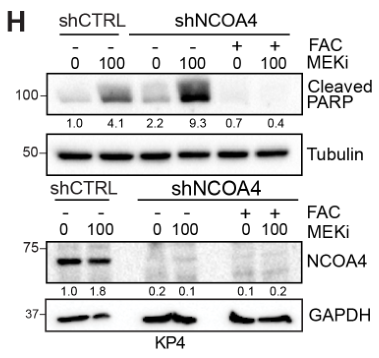
MEKi联合shNCOA4可诱导KP4细胞的凋亡标记物上调(Cleaved-PARP是细胞凋亡标志之一),但此效应可通过补充外源性铁阻断:
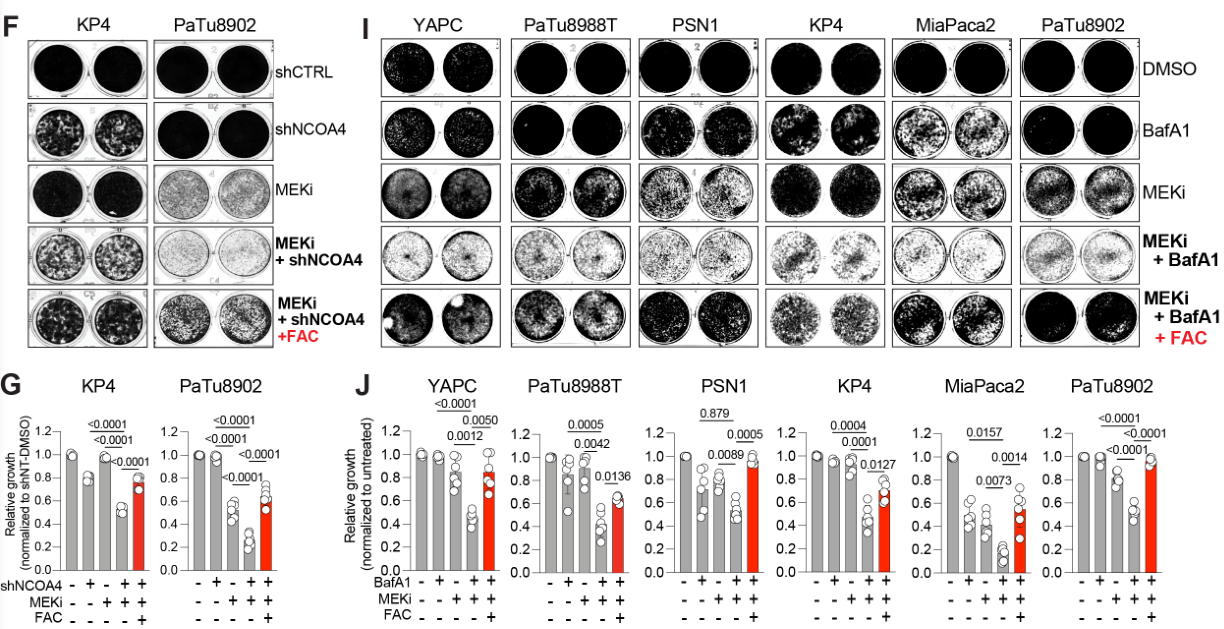
后续的平板克隆实验也进一步表明MEKi+shNCOA4介导细胞凋亡的效应可被外源性铁阻断:
应激后自噬-溶酶体特征广泛激活并与MYC活性负相关
在其它肿瘤的细胞株和数据集里面验证结论的普遍性
在肠癌细胞株和NSCLC细胞株中进一步验证MEKi可上调溶酶体基因并下调MYC靶基因:
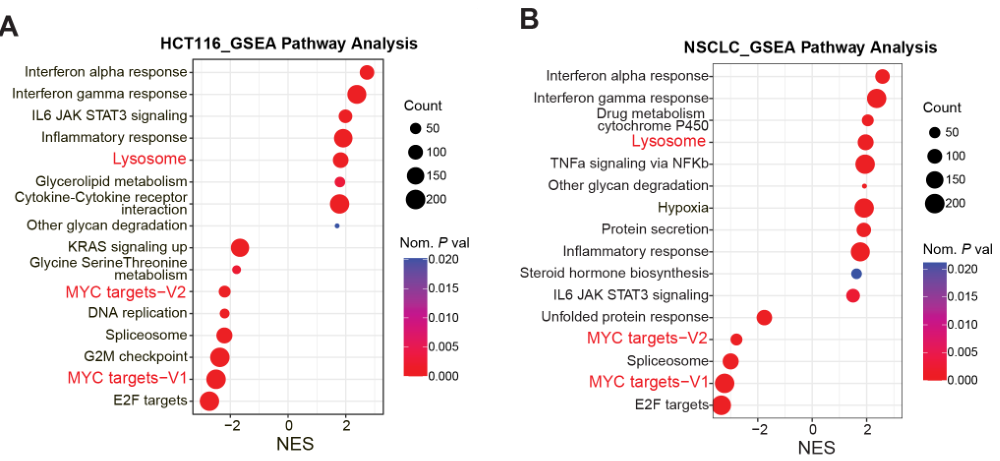
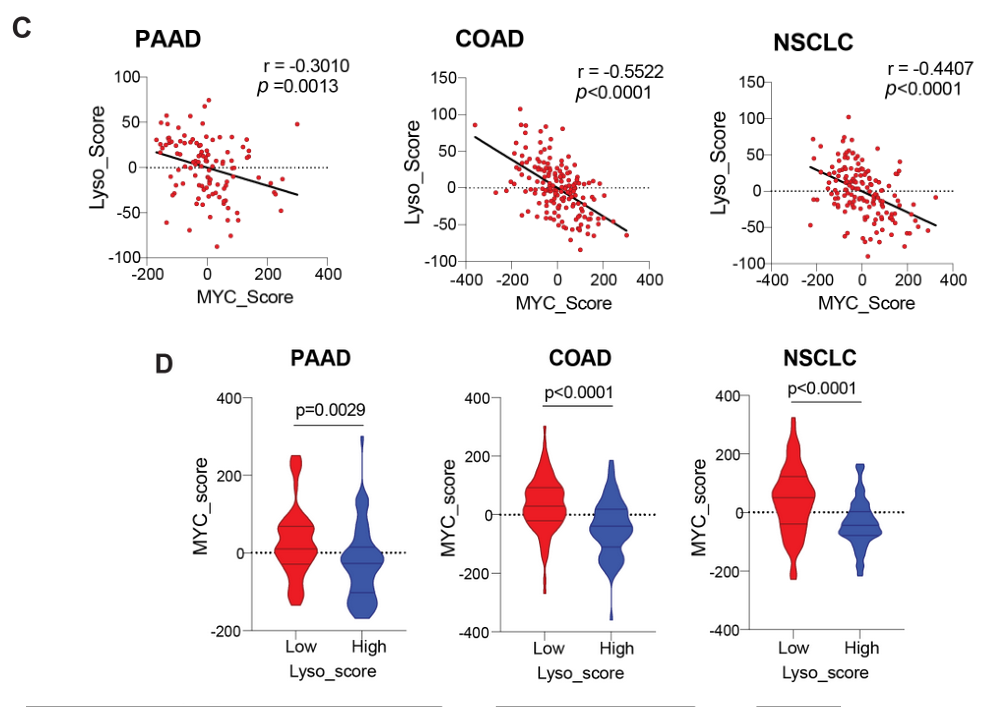
基于TCGA的胰腺癌、肠癌、肺癌数据库的分析表明MYC通路激活水平与溶酶体通路激活水平呈负相关:
小结
本研究的主要创新点在于发现MiT/TFE调控铁代谢在MEKi诱导自噬-溶酶体活性增强的过程中发挥重要作用。虽然还没看到文章的补充材料,但文章整体框架比较简单(至少比文献精读 第2期要简单1个数量级),也没有很复杂的动物实验。论点论证的过程中,实验设计比较简洁,并不着重通过控制多种混杂因素来增强论证;实验方法比较常规。作者发现MEKi诱导铁自噬的过程后,基于Fe2+——线粒体代谢的基本线索展开了后续MEKi对线粒体活性影响的相关研究,逻辑过程自然不突兀,也具有启发性。整体上,研究机制广度要比深度更加突出些。当然,作者最后也对研究结论在泛癌层面进行探究和讨论,不过只是浅尝辄止,这算是本研究中较弱的一环。
本研究最大的亮点是具有较强的临床转化能力。与《文献精读 第2期 靶向cGAS-STING-IL6治疗CIN肿瘤》类似,本文也提出了一种可行性很强的临床治疗策略——抑制自噬有望用于治疗KRAS/MEK/ERK抑制剂耐受的胰腺癌患者。无论是Trametinib还是氯喹都是FDA批准的药物(至少在临床试验的层面上),“Trametinib+氯喹”联合疗法或可让PDA患者获益。但本文又不止于此——通过表明铁自噬在MEKi耐药中的关键作用,为未来靶向铁自噬治疗KRAS/MEK/ERK抑制剂耐受胰腺癌患者奠定研究基础。也许这也是它凭借相对普通的实验论证却可以在Cancer Discov这种顶刊中发表的主要原因之一;从比较功利的角度上看的话,这是一个性价比很高的研究。
从最近看的两篇Nature大作,我发现它们的基调是创新点不多,但论证过程详尽,转化潜力巨大。当然,现实中不乏一些研究可能会一下子将前沿推进很远,但那种研究往往是可遇而不可求的。大多数的研究(甚至是Nature级别的研究)都是一点点推进科学发展和进步的,没有浮夸和冒进。相较于繁杂的机制探讨和论证,研究者贴近临床的“接地气”的朴实的研究思维让我更加印象深刻。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍

老哥,你是生物的吗,竟然还做文献阅读,我也是医学的
有关系吧 至于具体是啥暂不方便透露哈 有什么问题可以交流
大佬是啥专业的?