本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- Williams-Beuren综合征听觉增强机制
- 髓母细胞瘤分子亚型与菱形唇发育
- 一例快速射电暴的观察
- 核心结合因子α与后脑发育相关髓母细胞瘤
- CLN3与GPD的溶酶体积累
前言
最近在赶一审意见的response,所以文献看得少。以后会补上的!
本文是前沿快讯的第8期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。
CNS类
Williams-Beuren综合征听觉增强机制
Innate frequency-discrimination hyperacuity in Williams-Beuren syndrome mice. Cell
- Williams-Beuren 综合征 (WBS) 是一种由 27 个连续基因的半合子微缺失引起的罕见疾病。尽管存在神经发育和认知缺陷,但 WBS 患者的音乐和听觉能力得以保留或增强,这可能有助于深入了解听觉感知的遗传基础。
- 在这里,我们报告 WBS 的小鼠模型天生就增强了频率辨别敏锐度并改善了听觉皮层 (ACx) 中的频率编码。化学遗传学拯救显示频率辨别超敏是由 ACx 中过度兴奋的中间神经元引起的。一个 WBS 基因 Gtf2ird1 的单倍体不足通过下调神经肽受体 VIPR1 来复制 WBS 表型。 VIPR1 在 WBS 个体的 ACx 和源自具有 WBS 微缺失的人类诱导多能干细胞的脑类器官中减少。 ACx 中间神经元中的 Vipr1 缺失或过表达分别模拟或逆转了 WBS 小鼠的细胞和行为表型。
- 因此,ACx 中间神经元中的 Gtf2ird1-Vipr1 机制可能是 WBS 中卓越听觉敏锐度的基础。
髓母细胞瘤分子亚型与菱形唇发育
Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature
- 髓母细胞瘤是一种恶性儿童小脑肿瘤,在分子上分为不同的生物学亚组,这表明个性化的治疗方法将是有益的。小鼠建模和跨物种基因组学为离散的、亚组特异性发育起源提供了越来越多的证据。然而,发育中的人体组织的解剖学和细胞复杂性 3 – 特别是在菱形唇生发区内,它在内化到小脑结节之前产生所有谷氨酸能神经元谱系 – 使得难以验证先前从小鼠研究中得出的推论。
- 在这里,我们使用多组学来解决发育中的人类小脑中髓母细胞瘤亚群的起源。在人类菱形唇衍生谱系轨迹中编码的分子特征与第 3 组和第 4 组髓母细胞瘤中保持的感光器和单极刷细胞表达谱对齐,表明存在收敛基础。
- 对前瞻性机构队列的系统诊断影像学回顾将第 3 组和第 4 组肿瘤的假定解剖起源定位于结节。
- 我们的研究结果将具有临床挑战性的髓母细胞瘤亚群的分子和表型特征与其在人类发育早期阶段在菱形唇中的统一起点联系起来。
一例快速射电暴的观察
A fast radio burst source at a complex magnetized site in a barred galaxy. Nature
神奇的天文学
- 快速射电暴 (FRB) 是高度分散的毫秒级射电暴。最近对银河系 FRB的观测表明,至少有一些 FRB 起源于磁星,但宇宙学 FRB 的起源仍未确定。
- 在这里,我们报告了从重复源 FRB 20201124A在 54 天内在 82 h 内检测到 1,863 次爆发。这些观测显示法拉第旋转测量 (RM) 的不规则短时间变化,该测量检查前 36 天中单个爆发的密度加权视线磁场强度,然后是恒定的 RM。我们在超过一半的突发样本中检测到圆极化,其中一个突发达到 75% 的高分数圆极化。检测了分数线性和圆偏振中的振荡,以及作为波长函数的偏振角。所有这些特征都为在距源约一个天文单位内的复杂、动态演变、磁化的直接环境提供了证据。
- 我们对其银河系大小、富含金属的宿主星系的光学观测显示了一个棒螺旋,FRB 源位于中等星系中心距离的低恒星密度臂间区域。这种环境与在大质量恒星的极端爆炸中形成的年轻磁星引擎不一致,导致长时间的伽马射线爆发或超发光超新星爆发。
核心结合因子α与后脑发育相关髓母细胞瘤
Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature
有很多数据示例,值得收藏和研究。
- 髓母细胞瘤 (MB) 包括一组异质的后脑儿科胚胎性肿瘤,与后脑的早期发育密切相关。激活声波刺猬信号的突变导致上菱形唇 (RL) 颗粒细胞谱系中的Sonic hedgehog MB。相比之下,激活 WNT 信号的突变导致下 RL中的 WNT MB。然而,人们对更常见的组 4 (G4) MB 知之甚少,它被认为出现在单极刷状细胞谱系中。
- 在这里,我们证明导致 G4 MB 的体细胞突变集中在核心结合因子 α (CBFA) 复合物上,并且相互排斥的改变会影响 CBFA2T2、CBFA2T3、PRDM6、UTX 和 OTX2。 CBFA2T2 早期在智人小脑 RL 脑室下区的祖细胞中表达,而 G4 MB 在转录上类似于这些祖细胞,但在发育时间上停滞不前。在模型系统中敲除 OTX2 可缓解这种分化阻滞,从而使 MB 细胞沿着正常的发育分化轨迹自发进行。
- 分裂的人类 RL 的特殊性质,注定会产生人脑中的大部分神经元,其高水平的易感 EOMES+KI67+ 单极刷状细胞祖细胞可能使我们的物种易于发育 G4 MB。
CLN3与GPD的溶酶体积累
CLN3 is required for the clearance of glycerophosphodiesters from lysosomes. Nature
- 溶酶体具有许多作用,包括降解大分子和向细胞核发出信号。溶酶体功能障碍发生在各种人类疾病中,例如常见的神经退行性疾病和单基因溶酶体贮积症 (LSD)。对于大多数 LSD,致病基因已被确定,但在某些情况下,相关基因的功能是未知的,部分原因是溶酶体仅占细胞体积的一小部分,因此难以检测到溶酶体含量的变化。
- 在这里,我们开发了 LysoTag 小鼠,用于组织特异性分离与其内容物的多模式分析兼容的完整溶酶体。我们使用 LysoTag 小鼠研究 CLN3,这是一种功能未知的溶酶体跨膜蛋白。
- 在儿童中,CLN3 的缺失会导致幼年神经元蜡样脂褐质沉积症(巴顿病),这是一种致命的神经退行性 LSD。来自缺乏 CLN3 的小鼠大脑溶酶体的非靶向代谢物分析揭示了甘油磷酸二酯 (GPD) 的大量积累——甘油磷脂分解代谢的最终产物。 GPDs 也在 CLN3 缺陷培养细胞的溶酶体中积累,我们表明 CLN3 是其溶酶体出口所必需的。 CLN3 的丢失也会破坏溶酶体中的甘油磷脂分解代谢。
- 最后,我们发现 Batten 病患者的脑脊液中甘油磷酸肌醇水平升高,这表明甘油磷酸肌醇可能用作疾病生物标志物。
- 我们的研究结果表明,CLN3 是 GPD 的溶酶体清除所必需的,并且表明 Batten 病是一种神经退行性 LSD,具有甘油磷脂代谢缺陷。
LRR-RLPs与植物免疫
Plant receptor-like protein activation by a microbial glycoside hydrolase. Nature
- 植物依靠细胞表面定位的模式识别受体来检测病原体或宿主衍生的危险信号并触发免疫反应。具有富含亮氨酸重复 (LRR) 胞外域的受体样蛋白 (RLP) 构成模式识别受体的一个亚组,在植物免疫中发挥关键作用。 LRR-RLPs 的配体识别和激活机制仍然难以捉摸。
- 在这里,我们报告了来自本氏烟草的 LRR-RLP RXEG1 的晶体结构,该晶体结构识别来自病原体大豆疫霉的 XEG1 木葡聚糖酶。该结构显示特异性 XEG1 识别主要由 RXEG1 的氨基末端和羧基末端环出区域 (RXEG1(ID)) 介导。这两个环与 XEG1 的活性位点凹槽结合,抑制其酶活性并抑制本氏烟草的疫霉感染。 XEG1 的结合通过 RXEG1(ID) 和最后四个保守的 LRR 促进 RXEG1(LRR) 与 LRR 型共同受体 BAK1 的结合,从而触发 RXEG1 介导的免疫反应。 apo-RXEG1(LRR)、XEG1-RXEG1(LRR) 和 XEG1-BAK1-RXEG1(LRR) 的结构比较表明 XEG1 的结合诱导 RXEG1(ID) N 端区域的构象变化并增强结构灵活性RXEG1(LRR)的 BAK1 相关区域。
- 这些变化允许 RXEG1(ID) 的折叠切换以招募 BAK1(LRR)。我们的数据揭示了配体诱导的 LRR-RLP 与 BAK1 异二聚化的保守机制,并表明 LRR-RLP 在植物免疫中的双重功能。
黑色素瘤中的生长分层
A cellular hierarchy in melanoma uncouples growth and metastasis. Nature
似乎非常有趣,可以重点关注作者的论证思路
比利时癌症生物学中心-鲁汶分子癌症生物学实验室的Jean-Christophe Marine团队
- 尽管黑色素瘤因其高度的异质性和可塑性而臭名昭著,但细胞状态多样性(cell-state diversity)的起源和程度仍然知之甚少。同样,尚不清楚重叠或不同的黑色素瘤亚群是否支持生长和转移性传播。
- 在这里,通过结合小鼠遗传学、单细胞和空间转录组学、谱系追踪和定量建模,我们提供了肿瘤生长分层模型的证据,该模型反映了胚胎神经嵴细胞命运规范和分化背后的细胞和分子逻辑。
- 我们表明,致瘤能力与空间定位的血管周围生态位相关,这是一种通过内皮细胞建立的细胞间通讯途径获得的表型。与只有一小部分细胞注定会促进生长的模型一致,对具有间充质样状态的黑色素瘤细胞群的时间单细胞追踪显示,这些细胞不会促进原发性肿瘤的生长,而是构成一组转移起始细胞,它们在传播到次级器官时转换细胞身份。
- 我们的数据提供了黑色素瘤细胞状态多样性和轨迹的空间和时间分辨图,并表明支持生长和转移的能力仅限于不同的细胞池。
- 观察到这些表型能力可以在暴露于特定的生态位信号后动态获得,这保证了开发干扰这种微环境线索的癌细胞重编程活动的治疗策略。
蛋白水解靶向抗体
Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature
应用前景潜力巨大,可多留意
- 目前大多数靶向质膜受体的疗法通过拮抗配体结合或酶活性来发挥作用。然而,典型的哺乳动物蛋白质包含执行离散但协调活动的多个域。因此,一个结构域的抑制通常不完全抑制蛋白质的功能。事实上,靶向蛋白质降解技术,包括靶向蛋白水解的嵌合体1 (PROTACs),已经突出了靶向降解相对于抑制的临床重要优势。然而,以高亲和力结合两个靶标的异双功能化合物的产生是复杂的,特别是当需要口服生物利用度时。
- 在这里,我们描述了蛋白水解靶向抗体 (PROTAB) 的发展,该抗体将细胞表面 E3 泛素连接酶与跨膜蛋白结合在一起,导致体外和体内靶标降解。我们专注于锌和无名指 3 (ZNRF3),一种 Wnt 响应连接酶,并表明这种方法可以实现结直肠癌特异性降解。
- 值得注意的是,通过检查额外的细胞表面 E3 泛素连接酶和跨膜受体的基质,我们证明了该技术可用于“按需”降解。
- 此外,我们通过工程优化的抗体形式提供了关于控制靶标降解的基本规则的见解。
- 总之,这项工作描述了一种快速开发有效的、生物可利用的和组织选择性的细胞表面蛋白降解剂的策略。
免疫速率与抗体发生
Long-primed germinal centres with enduring affinity maturation and clonal migration. Nature
HIV疫苗相关的基础研究,很有趣
- 生发中心是抗体进化的引擎。在这里,我们在恒河猴中使用人类免疫缺陷病毒 (HIV) Env 蛋白免疫原引发,然后长时间不进一步免疫,我们证明了生发中心 B (BGC) 细胞可以持续至少 6 个月。与常规免疫相比,到第 10 周时,BGC 细胞增加了 186 倍。
- 单细胞转录分析表明,亮区和暗区生发中心状态都是持续的。 BGC 细胞的抗体体细胞超突变在整个 29 周的启动期持续积累,有选择压力的证据。在 29 周时,与 Env 结合的 BGC 细胞仍然比基线高 49 倍,这表明它们可以在更长时间内保持活跃。
- 单次加强免疫后产生高滴度的 HIV 中和抗体。完全糖基化的 HIV 三聚体蛋白是一种复杂的抗原,对 B 细胞 1,2 构成了相当大的免疫优势挑战。在这些长启动条件下产生的记忆 B 细胞具有更高水平的抗体体细胞超突变,并且记忆 B 细胞和抗体都更有可能识别非免疫优势表位。
- 鉴定了超过 6 个月生发中心期的大量 BGC 细胞谱系系统发育,证明了至少 191 天的持续生发中心活动和选择,没有进一步的抗原暴露。
- 一种长启动、缓慢递送(12 天)的免疫方法为难以实现的疫苗目标提供了希望,并表明耐心对于调整生发中心以最大限度地提高抗体反应具有重要价值。
COVID-19感染预后的遗传异质性
Common human genetic variants of APOE impact murine COVID-19 mortality. Nature
真香!此文再次证明,对科研来说,ideas和impact永远是第一位的。
- 严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 感染的临床结果高度异质,从无症状感染到 2019 年致命的冠状病毒病 (COVID-19)。这种异质性背后的因素仍未得到充分了解。遗传关联研究表明,遗传变异导致 COVID-19 结果的异质性,但对潜在的潜在因果机制了解不足。
- 在这里,我们展示了载脂蛋白 E (APOE) 基因的常见变体,在大约 3% 的世界人口中为纯合子 ,并与阿尔茨海默病、动脉粥样硬化和抗肿瘤免疫相关,在小鼠模型中影响 COVID-19 结果,概括如下:男性和高龄赋予的易感性增加。
- 与携带最普遍的 APOE3 等位基因的小鼠相比,携带 APOE2 或 APOE4 变体的小鼠表现出快速的疾病进展和较差的生存结果。 APOE2 和 APOE4 小鼠在感染后早期表现出病毒载量增加以及适应性免疫反应受到抑制。体外试验表明,相对于 APOE3,存在 APOE2 和 APOE4 的情况下感染增加,表明不同的结果是由 APOE 变体对病毒感染和抗病毒免疫的不同影响介导的。
- 与这些在小鼠体内的发现一致,APOE 基因型与英国生物库中 SARS-CoV-2 感染患者的存活率相关(候选变异分析,P = 2.6×10-7)。
- 我们的研究结果表明 APOE 基因型可以部分解释 COVID-19 结果的异质性,并需要进行前瞻性研究来评估 APOE 基因分型,以作为识别不良结果高风险患者的一种手段。
肿瘤免疫类
PERK抑制促进T细胞免疫激活
靶向肿瘤细胞激活免疫的研究范式,值得关注
- 在经历内质网应激的癌细胞中激活未折叠蛋白反应 (UPR) 可促进存活。然而,肿瘤细胞中的 UPR 如何影响抗肿瘤免疫反应的描述仍然很少。
- 在这里,我们研究了 UPR 介质胰腺 ER 激酶 (PKR) 样 ER 激酶 (PERK) 在癌细胞中在调节抗肿瘤免疫中的作用。删除癌细胞中的 PERK 或在荷黑色素瘤小鼠中对 PERK 进行药理学抑制会激发抗肿瘤 T 细胞免疫的强烈激活并减弱肿瘤生长。ER 应激恶性细胞中的 PERK 消除触发 SEC61β 诱导的下垂,从而促进免疫原性细胞死亡 (ICD) 和全身抗肿瘤反应。
- PERK 消融肿瘤中的 ICD 诱导刺激树突状细胞 (DC) 中 I 型干扰素的产生,这促使 CCR2 依赖性肿瘤运输常见的单核细胞前体,并将其在肿瘤内转化为单核细胞系炎性 Ly6C+CD103+ DCs。
- 这些发现确定了肿瘤细胞衍生的 PERK 如何促进免疫逃避,并突出了 PERK 靶向疗法在癌症免疫治疗中的潜力。
阻断MSP1-STING信号增强KL突变型肺癌ICB疗效
MPS1 inhibition primes immunogenicity of KRAS-LKB1 mutant lung cancer. Cancer Cell
- 由于内在的线粒体功能障碍,KRAS-LKB1 (KL) 突变型肺癌使 STING 沉默,导致 T 细胞排斥和对PD-1/PD-L1阻断的抵抗。
- 在这里,我们发现 KL 细胞还可以最大限度地减少 2’3′-环状 GMP-AMP (2’3′-cGAMP) 的细胞内积累,以进一步避免下游 STING 和 STAT1 激活。选择此漏洞的无偏筛选显示瞬时 MPS1 抑制 (MPS1i) 通过微核生成有效地重新参与 KL 细胞中的该途径。这种效应通过 STING 的表观遗传去抑制显着放大,并且只需要脉冲 MPS1i 治疗,与非分裂细胞相比,创造了一个治疗窗口。
- 单疗程的地西他滨治疗和脉冲 MPS1i 治疗可恢复体内 T 细胞浸润,增强抗 PD-1 功效,并产生持久反应,而没有显着毒性的证据。
IFNγ信号缺失可增强肿瘤ICB敏感性
In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol, IF2021=31.25
这个机制对于PAD-I型胃癌的解释有重要参考意义。
- 免疫系统可以消除肿瘤,但检查点可以使免疫逃逸。在这里,我们在使用免疫检查点阻断 (ICB) 治疗的癌症模型中使用基因组规模的体内 CRISPR 筛选来识别免疫逃避机制。
- 我们确定了在癌症中保守的免疫逃避基因和重要的免疫抑制检查点,包括非经典主要组织相容性复合物 I 类(MHC I 类)分子 Qa-1b/HLA-E。令人惊讶的是,肿瘤干扰素-γ (IFNγ) 信号传导的丧失使许多模型对免疫敏感。
- 肿瘤干扰素感应的免疫抑制作用是通过两种机制介导的。首先,经典 MHC I 类的肿瘤上调抑制自然杀伤细胞。其次,干扰素诱导的 Qa-1b 表达通过 ICB 诱导的 NKG2A/CD94 受体抑制 CD8+ T 细胞。
- 最后,我们表明强 IFN 特征与肾细胞癌或黑色素瘤患者对 ICB 的不良反应有关。这项研究表明,干扰素介导的经典和非经典 MHC I 类抑制检查点的上调可以促进免疫逃逸。
axi-cel与tisa-cel疗效的匹配比较研究
- Axicabtagene ciloleucel (axi-cel) 和 tisagenlecleucel (tisa-cel) 在复发/难治性 (R/R) 弥漫性大 B 细胞淋巴瘤 (DLBCL) 中均表现出令人印象深刻的临床活性。
- 在这项研究中,我们分析了 809 名 R/R DLBCL 患者在接受过两种或多种先前治疗后的结果,这些患者有商业嵌合抗原受体 (CAR) T 细胞订单用于 axi-cel 或 tisa-cel,并在回顾性法国 DESCAR-T 注册研究 (NCT04328298)。
- 在 1:1 倾向评分匹配 (n = 418) 后,与 tisa-cel 治疗的患者相比,最佳总反应率/完全反应率 (ORR/CRR) 为 80%/60%,而接受 axi-cel 治疗的患者为 66%/42%。 cel 分别为(ORR 和 CRR 比较的 P < 0.001)。中位随访 11.7 个月后,axi-cel 的 1 年无进展生存率为 46.6%,tisa-cel 为 33.2%(风险比 (HR) = 0.61;95% 置信区间 (CI),0.46 -0.79;P = 0.0003)。与输注 tisa-cel 后相比,输注 axi-cel 后的总生存期 (OS) 也显着提高(1 年 OS 63.5% 对 48.8%;HR = 0.63;95% CI,0.45-0.88;P = 0.0072)。使用治疗加权统计方法的逆概率观察到类似的结果。 axi-cel 组 1-2 级细胞因子释放综合征的发生率明显高于 tisa-cel,但≥3 级未观察到显着差异。关于免疫效应细胞相关神经毒性综合征 (ICANS),axi-cel 的 1-2 级和 ≥3 级 ICANS 明显高于 tisa-cel。
- 总之,我们的匹配比较研究支持在 R/R DLBCL 的第三个或更多治疗线中,axi-cel 与 tisa-cel 相比具有更高的功效和更高的毒性。
PCNSL外周体液的单细胞分析
Intratumor heterogeneity and T cell exhaustion in primary CNS lymphoma. Genome Med
思路其实蛮简单的,有点意思。但对于其它类型肿瘤研究的参考价值不高。
- 原发性中枢神经系统淋巴瘤(PCNSL)是一种罕见的中枢神经系统淋巴瘤,通常具有弥漫性大 B 细胞表型。立体定向活检和组织病理学检查是诊断标准。然而,从中枢神经系统活检中可获得的材料有限,因此阻碍了对 PCNSL 的深入表征。
- 我们对活检材料、血液和脑脊液 (CSF) 释放的 PCNSL 细胞进行流式细胞术、单细胞 RNA 测序和 B 细胞受体测序,并对活检样本进行空间转录组学。
- PCNSL 释放的细胞主要是活化的 CD19+CD20+CD38+CD27+ B 细胞。在单细胞RNA测序中,PCNSL细胞转录异质,形成多个恶性B细胞簇。过度扩增的 B 细胞克隆在活检和脑脊液之间共享,但不是血液衍生细胞。肿瘤微环境中的 T 细胞上调免疫检查点分子,从而识别来自 PCNSL 细胞的免疫逃避信号。空间转录组学揭示了恶性 B 细胞簇的异质空间组织,反映了它们在患者之间的转录异质性,以及 T 细胞衰竭标志物的明显表达,与高度恶性 B 细胞簇共定位。
- PCNSL 中的恶性 B 细胞显示出转录和空间肿瘤内异质性。 T 细胞耗竭在 PCNSL 微环境中很常见,与恶性细胞共定位,并突出了个性化治疗的潜力。
CXCL16/CXCR6信号与RNA病毒相关学习记忆缺陷
- 针对中枢神经系统 (CNS) 的新兴 RNA 病毒会导致幸存者出现认知后遗症。对感染西尼罗河病毒 (WNV) 的人类和小鼠的研究表明,西尼罗河病毒 (WNV) 是一种与学习和记忆缺陷相关的重新出现的 RNA 病毒,揭示了海马内小胶质细胞介导的突触消除。此外,CNS 驻留记忆 T (TRM) 细胞激活小胶质细胞,限制突触恢复并诱导 WNV 恢复小鼠的空间学习缺陷。参与 T 细胞-小胶质细胞相互作用的信号是未知的。
- 在这里,我们使用单细胞 RNA 测序检查了小鼠 WNV 恢复的前脑内的免疫细胞,以鉴定参与 T 细胞和小胶质细胞之间细胞间通讯的假定配体-受体对。聚类和差异基因分析之后是蛋白质验证以及基于遗传和抗体的方法,利用已建立的 WNV 恢复小鼠模型,其中小胶质细胞和补体促进持续的海马突触损失。
- 在小鼠感染后 25 天对宿主转录组免疫细胞的分析揭示了前脑稳态小胶质细胞向激活亚群的转变,这些亚群具有先前在神经退行性疾病研究中观察到的转录特征。重要的是,CXCL16/CXCR6 是一种参与 TRM 细胞生物学的趋化因子信号通路,被确定为在 WNV 恢复的前脑内严格调节表达 CD8+ TRM 细胞数量的 CXCR6。
- 我们证明 CXCL16 在所有骨髓细胞中高度表达,其独特的受体 CXCR6 在所有 CD8+ T 细胞上高度表达。使用遗传和药理学方法,我们证明 CXCL16/CXCR6 不仅是在感染后 CNS 中维持 WNV 特异性 CD8 TRM 细胞所必需的,而且还有助于它们的 TRM 细胞标志物的表达。此外,神经胶质激活和持续的突触消除需要 CXCR6+CD8+ T 细胞。
- 我们对 CXCL16/CXCR6 作为维持前脑 TRM 细胞、小胶质细胞和星形胶质细胞激活以及病毒恢复动物持续突触消除的小胶质细胞和 CD8+ T 细胞之间的相互作用联系的作用进行了全面评估。我们还表明,在小鼠恢复期间,CXCL16 的治疗靶向可能会减少 CNS CD8+ TRM 细胞。
lncRNA TGFB2-AS1
经典套路的lncRNA,在PNAS这种级别的杂志上还是很少见的
- 三阴性乳腺癌 (TNBC) 是最具挑战性的乳腺癌亚型,因为其复发率高、转移潜力大和总生存期短。癌细胞如何通过将非癌干细胞样细胞转化为具有干细胞特性的癌细胞来获得转移效力尚不清楚。
- 在这里,我们将长非编码 RNA (lncRNA) TGFB2-AS1 鉴定为 TNBC 中非癌干细胞群可逆性和可塑性的重要调节因子。我们发现 TGFB2-AS1 在体外会损害 TNBC 细胞的乳腺癌干细胞样细胞 (BCSC) 特征,并在体内显着降低致瘤频率和肺转移。
- 机制上,TGFB2-AS1 与 SWI/SNF 染色质重塑复合物的核心亚基 SMARCA4 相互作用,导致其靶基因(包括 TGFB2 和 SOX2)以顺式或反式方式转录抑制,从而抑制转化生长因子β(TGFβ)信号传导和 BCSC 特征。
- 与此一致,TGFB2-AS1 在原位 TNBC 小鼠模型中的过表达显着消除了 TGFβ2 赋予的肿瘤生长和肺转移的增强作用。此外,TNBC 患者 TGFB2-AS1 和 TGFβ2 的联合预后分析表明,高 TGFB2-AS1 和低 TGFβ2 水平与更好的结果相关。
- 这些发现证明了 TGFB2-AS1 基于改变 TNBC 的癌细胞命运在抑制 TNBC 疾病进展中的关键作用,并为 TNBC 患者的治疗提供了启示。
MDSCs亚群差异
Distinct cell adhesion signature defines glioblastoma myeloid-derived suppressor cell subsets. Cancer Res
比较有趣的研究,可以看看方法学
- 在多种类型的癌症中,髓源性抑制细胞 (MDSC) 的频率增加与较差的结果和较差的治疗反应有关。在胶质母细胞瘤 (GBM) 微环境中,单核 (m) MDSC 代表主要子集。然而,与粒细胞 (g) MDSC 相比,肿瘤微环境中 mMDSC 富集的分子基础尚未确定。
- 在这里,我们对 MDSC 亚群进行了第一次广泛的表观遗传分析,以定义潜在的细胞内在行为差异,并发现 mMDSCs 中细胞粘附程序的增强基因可及性与其在过继转移后 GBM 模型中的肿瘤加速能力有关。
- 作为增强细胞粘附特征的一部分,与 gMDSCs 相比,小鼠和人类 mMDSCs 表达更高水平的整合素 β1 和二肽基肽酶 4 (DPP-4)。整合素 β1 阻断消除了 mMDSC 的促肿瘤表型并改变了肿瘤微环境中的免疫特征,而用 DPP-4 抑制剂治疗延长了临床前 GBM 模型的生存期。
- 在 mMDSC 中靶向 DPP-4 可减少 pERK 信号传导及其向肿瘤细胞的迁移。
- 这些发现揭示了 MDSC 亚群分子基础的根本差异,并表明整合素 β1 和 DPP-4 代表了假定的免疫治疗靶点,以减弱 GBM 中骨髓细胞驱动的免疫抑制。
p-eIF2a活化与T细胞蛋白质翻译抑制
Stress-mediated attenuation of translation undermines T-cell activity in cancer. Cancer Res
- 蛋白质合成支持强大的免疫反应。肿瘤微环境 (TME) 中的营养竞争和全球细胞应激源可能会影响 T 细胞中的蛋白质翻译和抗肿瘤免疫。使用人类和小鼠肿瘤,我们在这里证明了 T 细胞中的蛋白质翻译在实体瘤中受到抑制。
- TME 中 T 细胞的葡萄糖可用性降低导致未折叠蛋白反应 (UPR) 元件 eIF2a 的激活。遗传小鼠模型显示,由活化的 p-eIF2a 介导的翻译衰减会破坏 T 细胞抑制肿瘤生长的能力。重编程 T 细胞代谢能够减轻 TME 中 p-eIF2a 的积累和翻译衰减,从而实现持续的蛋白质翻译。
- 代谢和药理学方法表明,蛋白酶体活性减轻了 p-eIF2a 的诱导,以支持最佳的抗肿瘤 T 细胞功能,防止翻译衰减并延长实体瘤中的细胞因子合成。
- 总之,这些数据确定了一种新的治疗途径,以促进肿瘤免疫疗法的功效。
H3.3-G34R/V突变与cGAS-STING激活
H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J Clin Invest
- 小儿高级别胶质瘤 (pHGGs) 是美国儿童癌症相关死亡的主要原因。 16% 的半球儿科和年轻成人 HGG 编码组蛋白 H3.3 (H3.3-G34R/V) 中的 Gly34Arg/Val 替代。 H3.3-G34R/V 在 pHGGs 中驱动恶性肿瘤和治疗耐药的机制仍然未知。
- 使用同基因的基因工程小鼠模型 (GEMM) 和编码 H3.3-G34R 的人类 pHGG 细胞,我们证明H3.3-G34R/V突变导致 DNA 修复途径的下调。这导致对 DNA 损伤的敏感性增强和 DNA 损伤反应 (DDR) 的抑制。我们证明了 G34R 突变体 pHGG 中不正确的 DNA 修复导致的遗传不稳定性导致染色体外 DNA 的积累,从而激活 cGAS-STING 途径,诱导免疫刺激细胞因子的释放。
- 我们用放射疗法 (RT) 和 DNA 损伤反应抑制剂 (DDRi)(即血脑屏障可渗透 PARP 抑制剂、pamiparib 和细胞周期检查点 CHK1/2 抑制剂)组合治疗 H3.3-G34R pHGG 小鼠, AZD7762), 这些组合导致大约 50% 的长期幸存者。此外,添加 STING 激动剂 (diABZ1) 增强了这些治疗的疗效。长期幸存者产生了免疫记忆,在再次攻击时阻止了 pHGG 的生长。
- 这些结果表明,DDRi 和 STING 激动剂与 RT 联合使用可在 G34 突变体 pHGG 中诱导免疫介导的治疗效果。
Fas decoy与实体肿瘤的免疫激活
Secreted Fas decoys enhance the antitumor activity of engineered and bystander T cells in Fas ligand-expressing solid tumors. Cancer Immunol Res
- T 细胞免疫疗法已在某些血液系统恶性肿瘤中显示出显着的临床效果。然而,实体瘤的疗效一直欠佳,部分原因是由免疫抑制分子组成的敌对肿瘤微环境。一种在实体瘤中大量表达的抑制剂是 Fas 配体 (FasL),它可以触发表达 Fas 的效应细胞如 T 细胞和自然杀伤 (NK) 细胞的凋亡。
- 为了减轻 FasL 诱导的实体瘤中肿瘤特异性免疫细胞的抑制,我们在此描述了 Fas 诱饵的开发,该诱饵由工程细胞在激活后分泌并隔离配体,防止其与效应器表面的 Fas 结合细胞。我们通过创建 Fas 诱饵和 IL-15 细胞因子融合蛋白进一步改善了这种方法的免疫刺激效果,这增强了诱饵工程以及旁观者嵌合抗原受体 (CAR) T 细胞在异种移植物中的持久性和抗肿瘤活性胰腺癌模型。
- 我们的数据表明,分泌的 Fas 诱饵可以增强过继转移和内源性肿瘤特异性效应细胞在表达 FasL 的实体瘤中的功效。
TLS TIM4+巨噬细胞与免疫激活
A population of TIM4+FOLR2+ macrophages localized in tertiary lymphoid structures correlates to an active immune infiltrate across several cancer types. Cancer Immunol Res
蛮有意思的,转给YJS看一下
- TIM4 以前与抗肿瘤免疫有关,但这种受体在人类癌症组织中的表达模式和功能仍然没有得到很好的探索。在这里,我们将人体组织的广泛免疫标记与泛癌转录组数据集的计算机分析相结合,以探索 TIM4 表达的临床意义。
- 我们的结果揭示了 TIM4 在癌症患者的一小部分腔巨噬细胞 (CATIM4+MΦ) 上表达。此外,我们发现 TIM4 在癌相关TLS (TLS TIM4+MΦ) 的 T 细胞区的巨噬细胞上高表达。对泛癌数据集的计算机分析显示,TIM4 表达与 B 细胞标志物、效应 CD8+ T 细胞和定义三级淋巴结构的 12 趋化因子特征之间呈正相关。此外,TLSTIM4+MΦ 在微卫星不稳定性和高 CD8+ T 细胞浸润的癌症中富集,证实了它们与免疫反应性肿瘤的关联。 CATIM4+MΦ 和 TLSTIM4+MΦ 都表达 FOLR2,一种组织驻留 MΦ 的标记。
- 然而,与 TLSTIM4+MΦ 相比,CATIM4+MΦ 具有更高的免疫抑制分子 TREM2、IL10 和 TGFβ 的表达。通过分析肿瘤相关骨髓细胞的 scRNA-seq 数据集,我们确定了两个与 CATIM4+MΦ 和 TLSTIM4+MΦ 相干的 TIM4+FOLR2+ 簇。我们为每个子集定义了特定的基因特征,发现 CATIM4+ MΦ 特征与较差的患者生存率相关。相比之下,TLSTIM4+MΦ 基因特征与更好的预后呈正相关。
- 这些数据共同表明,TIM4 标志着两个不同的巨噬细胞群,具有不同的表型和组织定位,并且可能在肿瘤免疫中具有相反的作用。
抗VEGF联用丙咪嗪治疗胶质母细胞瘤
Cancer cell autophagy, reprogrammed macrophages, and remodeled vasculature in glioblastoma triggers tumor immunity. Cancer Cell
非常朴素的研究
- 胶质母细胞瘤 (GBM) 对治疗的反应很差并且总是致命的。规避这种难治性的一种可能的策略是共同针对疾病的独特机制成分,旨在同时破坏肿瘤进展和治疗抵抗所需的多种能力。
- 我们通过将重塑肿瘤脉管系统的血管内皮生长因子 (VEGF) 通路抑制剂与三环类抗抑郁药丙咪嗪结合来评估这一概念,丙咪嗪可增强 GBM 癌细胞的自噬,并通过抑制组胺受体信号传导意外地重新编程免疫抑制性肿瘤相关巨噬细胞,使其成为免疫刺激剂
- 虽然这两种药物都不能作为单一疗法有效,但丙咪嗪与 VEGF 通路抑制剂的组合可协调 CD8 和 CD4 T 细胞的浸润和活化,在几种 GBM 小鼠模型中产生显着的治疗益处。在最终复发的肿瘤中加入抗程序性死亡配体 1 (PD-L1) 的免疫检查点阻断可显着延长生存获益。
- 结果说明了机制指导的治疗共同靶向肿瘤微环境中不同生物脆弱性的潜力。
肿瘤转移类
MAD-9-NF-κB-Spp1信号的肝细胞特异性激活与肿瘤侵袭性
研究方法十分有趣,值得关注。
- 癌基因黑色素瘤分化相关基因 9/syndecan 结合蛋白 (MDA-9/SDCBP) 在许多促进侵袭性转移性疾病的癌症中过度表达。然而,MDA-9 在调节肝细胞癌 (HCC) 中的作用尚未得到充分研究。
- 为了揭示 MDA-9 在 HCC 中的功能,我们生成并表征了具有肝细胞特异性过表达 MDA-9 (Alb/MDA-9) 的转基因小鼠。与 WT 同窝小鼠相比,Alb/MDA-9 小鼠表现出显着更高的 N-亚硝基二乙胺/苯巴比妥诱导的 HCC 发生率,巨噬细胞显着活化和浸润。
- 幼稚 WT 和 Alb/MDA-9 肝细胞中的 RNA-seq 鉴定了与侵袭、血管生成和炎症相关的信号通路的激活,尤其是 NF-κB 和整合素相关激酶 (ILK) 信号通路。从幼稚肝脏纯化的非实质细胞中的 scRNA-seq 显示 Alb/MDA-9 小鼠与 WT 相比 Kupffer 细胞和巨噬细胞的活化。在 MDA-9 过表达后观察到分泌型磷蛋白 1 (Spp1/骨桥蛋白) 表达的强烈增加。 NF-κB 通路的抑制阻断了MDA-9诱导的Spp1表达,并且 Spp1 的敲低导致了 MDA-9 诱导的巨噬细胞迁移和血管生成的抑制。
- Alb/MDA-9 是第一个在任何组织类型中都有 MDA-9 过表达的小鼠模型。
- 我们的研究结果揭示了由 NF-κB 和 Spp1 介导的 MDA-9 促进 HCC 的新作用,并支持使用 MDA-9 抑制剂作为侵袭性 HCC 的潜在治疗方法的基本原理。
临床类
f-FTD症状发作预测
Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med
这个研究作者列表好长啊
- 与家族性阿尔茨海默病不同,我们无法准确预测症状前家族性额颞叶痴呆 (f-FTD) 突变携带者的症状发作,这是设计疾病预防试验的主要障碍。
- 我们开发了用于 f-FTD 疾病进展的多模式模型,并估计了 C9orf72、GRN 和 MAPT 突变携带者的临床试验样本量。模型包括 796 名携带者和 412 名非携带者对照的纵向临床和神经心理学评分、区域脑容量和血浆神经丝轻链 (NfL)。我们发现临床和生物标志物进展的时间顺序因基因型而异。在使用基于模型的患者选择的预防试验模拟中,萎缩和 NfL 是最佳终点,而临床测量是早期症状试验的潜在终点。 f-FTD 预防试验是可行的,但可能需要全球招募工作。
- 这些疾病进展模型将促进 f-FTD 临床试验的规划,包括选择最佳终点和入组标准,以最大限度地提高检测治疗效果的能力。
血清CRP与癌症风险
C-reactive protein and cancer risk: a pan-cancer study of prospective cohort and Mendelian randomization analysis. BMC Med, IF2021=11.15, Q1
很有趣的研究,方法学多多学习。CRP都能被您玩出花来!
- 背景:尽管观察性研究报告了血清 C 反应蛋白 (CRP) 浓度与肺癌、乳腺癌和结直肠癌风险之间的关联,但其他癌症的证据不一致或缺失。我们进行了泛癌分析,以全面评估 CRP 的作用,包括线性和非线性关联。
- 方法:我们分析了来自英国生物银行队列的 420,964 名无癌症参与者。进行了多变量调整的 Cox 比例风险模型,以评估观察到的 CRP 与总体癌症和 21 个部位特异性癌症风险的相关性。此外,我们进行了线性和非线性孟德尔随机化分析,以探索它们之间的潜在因果关系。
- 结果:在 7.1 年的中位随访期间(四分位距:6.3、7.7),观察到 34,979 例癌症病例。观察性分析表明,较高的 CRP 浓度与总体癌症风险增加相关(风险比 (HR) = 1.02,95% CI:1.01,每增加 1mg/L 为 1.02,P < 0.001)。 CRP 与总体癌症风险之间存在非线性关联,拐点为 3mg/L(错误发现率调整(FDR 调整)Poverall < 0.001 和 FDR 调整 Pnon-linear < 0.001)。对于特定部位的癌症,我们观察到食管癌和胃癌的正线性相关性(FDR 调整的 Poverall < 0.050 和 FDR 调整的 Pnon-linear > 0.050)。
- 此外,我们还观察到三种不同的非线性关联模式,包括“快速到低增长”(头颈癌、结直肠癌、肝癌、肺癌、肾癌和非霍奇金淋巴瘤)、“从快速到低增长”减少”(乳腺癌)和“减少到平台”(慢性淋巴细胞白血病)。此外,非线性关联模式的拐点始终在 3mg/L 左右。相比之下,没有证据表明遗传预测的 CRP 与整体癌症或特定部位癌症的风险之间存在线性或非线性关联。
- 结论:我们的结果表明,CRP 是评估整体癌症和 12 种特定部位癌症风险的潜在生物标志物,而未观察到遗传预测的 CRP 和癌症风险之间的关联。
乳腺癌预后GEP的临床价值
看看cut-off策略;临床低风险的标准是基于TNM Stage吗?
- 简介:预后基因表达特征可与经典临床病理学因素结合使用,以指导 ER 阳性、HER2 阴性乳腺癌的辅助化疗决策。然而,在治疗决策过程中引入基因组检测后的长期结果数据是有限的。
- 方法:在前瞻性 RASTER 研究中,对 427 名 cTanyN0M0 乳腺癌患者的肿瘤进行了测试,以评估 70 基因特征 (MammaPrint)。将结果提供给他们的治疗医师,以将其纳入辅助全身治疗的决策中。在这里,我们报告了 310 名 ER 阳性、HER2 阴性肿瘤患者在中位随访 10.3 年的临床和基因组风险类别的长期结果。
- 结果:在临床高危患者中,45 名 (49%) 被归类为基因组低危患者。在该亚组中,10 年时,接受 (95.7% [95% CI 87.7-100]) 和未接受 (95.5% [95% CI 87.1-100]) 化疗的患者之间的无远处复发间隔 (DRFI) 相似。在临床低风险患者组中,56 名 (26%) 被归类为基因组高风险。在临床低风险组中,超过 5 年,基因组高风险和低风险亚组之间出现差异,导致 10 年 DRFI 分别为 84.3% (95% CI 74.8-95.0) 和 93.4% (95% CI 89.5-97.5),分别。有趣的是,基因组超低风险患者的 10 年 DRFI 为 96.7% (95% CI 90.5-100),大部分 (79%) 没有全身治疗。
- 结论:这些数据证实,临床高风险、基因组低风险的肿瘤在共享决策的现实世界环境中具有出色的结果。连同 MINDACT 试验的更新结果,这些数据支持在 ER 阳性、HER2 阴性、淋巴结阴性、临床高危乳腺癌患者中使用 MammaPrint。
乳腺癌老年女性化疗衰弱相关炎症biomarkers
- 老年乳腺癌幸存者在辅助化疗后临床衰退的风险增加。本研究旨在评估辅助化疗前评估的炎症标志物是否与适合患有乳腺癌的老年人群中化疗引起的临床下降相关。
- 在一项对年龄 ≥ 65 岁且接受化疗的 I-III 期乳腺癌女性进行的前瞻性研究中,我们测量了白细胞介素 6 (IL-6) 和 C 反应蛋白 (CRP) 化疗前 (T1)。我们在 T1 和化疗后 (T2) 使用赤字累积指数 (DAI;分类为健壮、衰弱和衰弱) 评估虚弱状态。感兴趣的人群是 T1 时强壮的女性。主要结果是化疗引起的衰弱状态下降,定义为 DAI 从稳健 (T1) 下降到衰弱前或衰弱 (T2)。多变量逻辑回归用于检查炎症标志物与主要结果之间的关联,并根据社会人口学和临床特征进行调整。
- 在 T1 的 295 名健壮女性中,76 名 (26%) 经历了化疗引起的虚弱状态下降,其中 66% 的 IL-6 高,63% 的 CRP 高,46% 的 IL-6 和 CRP 高在 T1。在调整了社会人口学和临床特征后,与低 IL-6 和 CRP 的女性相比,高 IL-6 和 CRP 的女性有 > 3 倍(优势比 = 3.52;95% CI,1.55 至 8.01;P = 0.003)的几率化疗引起的衰弱状态下降。
- 在化疗开始前临床健康的老年早期乳腺癌女性队列中,高 IL-6 和 CRP 化疗前化疗与化疗引起的虚弱状态下降相关,与社会人口学和临床危险因素无关。需要进一步的研究来检验炎症标志物是否可以为治疗老年乳腺癌幸存者提供更个性化的方法。
CRC的同步肝转移瘤的切除时机与KRAS突变
Association of Simultaneous vs Delayed Resection of Liver Metastasis With Complications and Survival Among Adults With Colorectal Cancer. JAMA Netw Open
- 同时或延迟切除原发性结直肠癌 (CRC) 的同步肝转移瘤 (SLM) 仍然是一个有争议的话题。探讨可切除 SLM 患者同时切除与延迟切除的结果。这项比较有效性研究包括 2000 年 1 月 1 日至 2019 年 12 月 31 日期间在中国 3 个独立中心接受治愈性肝切除术的 1569 名可切除 SLM 患者。1:1 倾向评分匹配进行了。随访于 2021 年 8 月 31 日完成,数据分析时间为 2022 年 4 月 1 日至 30 日。
- 主要结果是术后 60 天内发生至少 1 种主要并发症的患者百分比。次要结局是术中和术后并发症、总生存期 (OS) 和癌症特异性生存期 (CSS) 率。
- 在纳入的 1569 名患者中,1057 名(67.4%)接受了延迟切除术(719 名男性 [68.0%],平均 [SD] 年龄为 57.4 [11.2] 岁),512 名患者(310 名男性 [60.5%]平均 [SD] 年龄 57.1 [10.5] 岁)接受了同步切除术。匹配产生了 495 对同时切除的患者。主要围手术期并发症的百分比在同时切除组和延迟切除组之间没有差异(34.1% vs 30.0%;P = .89)。延迟切除组的 3 年 OS 率为 65.2%,5 年为 47.1%,8 年为 38.0%,3 年为 78.0%,5 年为 65.4%,8 年为 63.1%。组(风险比 [HR],1.42;95% CI,1.10-1.85,P = .003)。延迟切除组 3 年 CSS 率为 68.3%,5 年为 48.5%,8 年为 37.1%,3 年为 79.2%,5 年为 67.2%,8 年为 65.9%。组(HR,1.45;95% CI,1.14-1.98;P = .004)。
- 在根据 KRAS 序列变异状态比较两种策略的亚组分析中,OS 率(HR,1.61;95% CI,1.45-2.18;P < .001)和 CSS 率(HR,1.62;95 CI,1.40-1.87) ; P = .003]) 在 KRAS 野生型肿瘤患者中,同时切除组明显优于延迟切除组。
- 结论和相关性:本研究结果表明,同时切除 CRC 和 SLM 时并发症发生率没有差异,同时切除的生存益处仅限于 KRAS 野生型肿瘤患者。将分子特征整合到治疗决策中是准确、个性化治疗的基础。
ICB与继发性糖尿病
- 免疫检查点抑制剂 (ICI) 已成为一种有前途的癌症治疗方法。然而,ICI 的使用会诱发免疫相关的不良事件,包括糖尿病。我们比较了接受 ICI 和接受常规化疗 (CC) 的患者新发糖尿病的风险。
- 使用三级医院数据库,我们纳入了既往无糖尿病病史且接受 CC 或 ICI 治疗的癌症患者。应用了一对五最近邻倾向匹配,并使用 Cox 比例风险模型估计了患糖尿病的风险。潜在类别增长建模是使用轨迹方法进行的,以确定随时间推移遵循相似葡萄糖轨迹模式的不同簇。
- 1326 名受试者中,1105 名接受 CC,221 名接受 ICI。 ICI 组新发糖尿病的风险显着高于 CC 组(调整后的风险比为 2.454,95% 置信区间为 1.528-3.940;p < 0.001)。与 CC 组相比,ICI 组在具有增加的葡萄糖模式的轨迹簇中具有更高比例的受试者(分别为 10.4 % 和 7.4 %)。在 ICI 组中,葡萄糖模式增加的受试者主要是男性,并且在 ICI 给药后与增强的淋巴细胞增多有关。
- 与 CC 相比,ICI 治疗与糖尿病发病风险增加有关。需要定期监测接受 ICI 治疗的患者的血糖水平,尤其是男性和 ICI 治疗后淋巴细胞增多的患者,以尽早发现 ICI 相关糖尿病。
急性缺血性脑卒中合并癌症与EVT术
Investigating Outcomes Post Endovascular Thrombectomy in Acute Stroke Patients With Cancer. Neurology.
编辑评论:Thrombectomy for Acute Stroke in People With Cancer: Hopes and Challenges. Neurology
- 癌症是急性缺血性卒中(AIS)患者的常见合并症。将血管内血栓切除术 (EVT) 确立为大血管闭塞治疗标准的随机对照试验通常将癌症患者排除在外。因此,目前尚不清楚血管内血栓切除术在癌症人群中的临床益处。
- 使用大型住院患者数据库,即国家住院患者样本 (NIS),检查接受 EVT 的癌症患者的临床结果。查询NIS 2016-2019年AIS入院情况,确定癌症患者。比较了患有和不患有癌症的 AIS 患者的基线人口统计学、合并症、再灌注治疗和结果。对于接受 EVT 的患者,倾向评分匹配用于研究主要结果,例如颅内出血风险、住院时间和出院处置。
- 在研究期间,2,677,200 名患者因 AIS 住院,其中 228,800 人(8.5%)被诊断为癌症。 132,210 名患者接受了 EVT,其中 8935 名 (6.8%) 患有癌症。超过 20% 的接受 EVT 的癌症患者在没有服务的情况下常规出院回家的良好结果。在调整倾向评分分析中,接受 EVT 的癌症患者的颅内出血发生率相似(OR 1.03,CI 0.79-1.33,p=0.90)和出院回家几率显着高于 10 天以上的长期住院率(或 1.34,CI 1.07-1.68,p=0.01)。与没有癌症的患者相比,接受 EVT 的转移性癌症患者的颅内出血发生率(OR 1.03,CI 0.64-1.67,p=1.00)和常规出院的可能性(OR 0.83,CI 0.51-1.35,p=0.54)相似)但住院死亡率较高 (OR 2.72, CI 1.52-4.90, p<0.01)。
- 我们的研究结果表明,在当代医疗实践中,合并癌症或转移性癌症的急性卒中患者接受血管内血栓切除术的颅内出血率和出院率与非癌症患者相似。这表明符合再灌注治疗标准的 AIS 患者可能会被考虑用于合并癌症的诊断。
乳腺癌易感基因
- 乳腺癌具有重要的遗传基础,其中约 60% 仍无法解释。检测 BRCA1/BRCA2 可以有效区分家庭中的乳腺癌风险,鉴定其他乳腺癌易感基因可以提供临床实用性。
- 我们纳入了通过 BOCS 研究招募的 2,135 例浸润性乳腺癌病例,这是一项英国家族性乳腺癌的回顾性研究。资格标准:女性,BRCA 阴性,欧洲白人种族,以及以下之一:i) 乳腺癌家族史,ii) 双侧疾病,iii) 发病年龄年轻(<30 岁),iv) 伴随卵巢癌。我们对病例进行外显子组测序,并对来自 gnomAD 的 51,377 个种族匹配的人群对照的罕见破坏性变异进行基因水平负荷测试。
- 159/2135 (7.4%) 例患者在已确定的乳腺癌易感基因中有合格变异,而其他癌症易感基因中的信号证据很少。已知的乳腺癌易感基因 PALB2、CHEK2 和 ATM 是唯一在多次测试校正后保持统计学显着性的基因。由于该系列中遗传病例的丰富性,我们有很好的能力 (>80%) 检测到 BRCA1 样风险基因 (优势比 = 10.6) 低至 4.6 x 10-5 (1在 10,799 人中,不到 BRCA1 的十分之一)和 PALB2 样风险(优势比 = 5.0)下降到 2.8 x 10-4 的人群次要等位基因频率(1,779 人中有 1 个,不到 PALB2 的一半)。鉴定新的中等外显率基因(优势比 = 2-3)如 CHEK2 和 ATM 的能力较低。
- 这是迄今为止发表的关于富集乳腺癌的最大病例对照全外显子组分析。虽然可能存在其他乳腺癌易感基因,但高外显率的基因可能具有非常低的突变频率。关于这些基因的临床效用存在争议。
PRISM项目利用液体活检检测活动性突变
研究方法了解一下。
暂无摘要。
其它类
染色体外DNA的动态演化
Deciphering the evolutionary dynamics of extrachromosomal DNA in human cancer. Nat Genetics
暂无描述。
综合培养基数据库
MediaDive: the expert-curated cultivation media database. Nucleic Acids Res
看摘要似乎有点水
- 我们展示了 MediaDive (https://mediadive.dsmz.de),这是一个由专家策划的综合培养基数据库,其中包含超过 3200 种标准化培养基的配方、说明和分子组成,适用于来自所有生命领域的超过 40 000 种微生物菌株.
- MediaDive 旨在实现广泛的应用,从研究和诊断实验室的日常使用到新媒体设计的知识驱动支持和人工智能驱动的数据挖掘。它提供了许多直观的搜索功能和比较工具,例如识别相关分类群的培养基和整合菌株特异性修饰。
- 除了经典的 PDF 存档和打印之外,最先进的网站还允许在移动设备上无纸化使用媒体食谱,方便湿实验室使用。此外,可以使用 RESTful Web 服务检索数据以进行大规模数据分析。内部编辑器界面确保来自莱布尼茨研究所 DSMZ 的培养专家不断扩展和管理媒体,这与 DSMZ 不断增长的微生物馆藏相互关联。外部用户参与由专用的媒体构建器工具覆盖。标准化和程序化可访问的数据将促进培养基设计的新方法,以针对绝大多数未培养的微生物。
lncRNA-Smad7调控Nodal/TGF-β 和 BMP 信号传导
LncRNA-Smad7 mediates cross-talk between Nodal/TGF-β and BMP signaling to regulate cell fate determination of pluripotent and multipotent cells. Nucleic Acids Res
- 转化生长因子 β (TGF-β) 超家族蛋白是细胞发育和分化的有效调节剂。 Nodal/Activin/TGF-β 和 BMP 配体在早期发育过程中都存在于细胞内和细胞外环境中,这两个发育信号分支之间的交叉对话是目前研究的热点。
- 在这里,我们展示了 Nodal 诱导的 lncRNA-Smad7 通过抑制小鼠胚胎干细胞 (mESCs) 中的 BMP 信号传导来调节细胞命运决定。 lncRNA-Smad7 的消耗显着损害了 mESCs 中的心肌细胞分化。
- lncRNA-Smad7 通过形成 R 环的 (CA)12 重复序列与 Bmp2 启动子区域结合来抑制 Bmp2 表达。重要的是,Bmp2 敲低挽救了由 lncRNA-Smad7 敲低诱导的心肌细胞分化缺陷。因此,lncRNA-Smad7 拮抗 mESC 中的 BMP 信号传导,并类似地调节 C2C12 小鼠成肌细胞中骨细胞和肌细胞形成之间的细胞命运决定。
- 此外,lncRNA-Smad7 与 mESCs 中的 hnRNPK 相关,并且 hnRNPK 在 Bmp2 启动子处结合,可能有助于 Bmp2 表达抑制。
- 本工作中描述的通过 lncRNA-Smad7 的 Nodal/TGF-β 和 BMP 信号传导之间的拮抗作用为理解早期发育中的细胞命运决定提供了一个框架。
细胞因子受体靶向嵌合体 (KineTACs)
Modular cytokine receptor-targeting chimeras for targeted degradation of cell surface and extracellular proteins. Nat Biotechnol. full html
- 通过溶酶体递送靶向降解细胞表面和细胞外蛋白是调节细胞外生物学的重要手段。然而,由于缺乏模块化、易于开发、受限的组织靶向以及对细胞表面和细胞外蛋白的适用性,这些方法具有局限性。
- 我们描述了一种溶酶体降解策略,称为细胞因子受体靶向嵌合体 (KineTACs),它解决了这些限制。 KineTAC 是完全基因编码的双特异性抗体,由结合其同源细胞因子受体的细胞因子臂和目标蛋白质的靶标结合臂组成。
- 我们表明,含有细胞因子 CXCL12 的 KineTAC 可以使用诱饵回收受体 CXCR7 将多种靶蛋白靶向溶酶体进行降解。其他 KineTAC 旨在利用其他 CXCR7 靶向细胞因子 CXCL11 和 vMIPII,以及白细胞介素 2 (IL-2) 受体靶向细胞因子 IL-2。
- 因此,KineTAC 代表了一种通用的、模块化的、选择性的和简单的遗传编码策略,用于诱导具有广泛或组织特异性分布的细胞外和细胞表面靶标的溶酶体递送。
细胞区域指纹与单细胞跟踪和谱系重建
Cell region fingerprints enable highly precise single-cell tracking and lineage reconstruction. Nat Method;感觉蛮有趣的,可以了解下。还有评论文章:Improved tracking via cell region fingerprints. Nat Methods
- 通过显微镜对细胞生长、遗传及其相关过程的实验研究需要足够持续时间的准确单细胞观察来重建谱系。然而,细胞追踪——将连续图像上的相同细胞分配给一个轨道——通常具有挑战性,导致人工验证费力。
- 在这里,我们提出了指纹来快速识别有问题的分配。指纹距离比较傅里叶变换低频中包含的结构信息,以测量两个连续图像中细胞之间的相似性。我们表明,只要图像具有足够的结构信息,指纹就可以广泛适用于细胞类型和图像模式。
- 我们的跟踪器 (TracX) 使用指纹来拒绝不太可能的分配,从而提高对已发布和新生成的长期数据集的跟踪性能。
- 对于酿酒酵母,我们提出了一个以 Whi5 调节器为中心的单细胞和群体水平的细胞大小控制综合模型,展示了精确跟踪如何帮助揭示以前未描述的单细胞生物学。
多肽动态作用的荧光显影
A fluorescent sensor for real-time measurement of extracellular oxytocin dynamics in the brain. Nat Methods
看上去很特别,有机会全文了解
- 催产素 (OT) 是一种下丘脑神经肽,可作为大脑中的神经调节剂,协调各种动物行为。然而,大脑 OT 动力学与复杂动物行为之间的关系在很大程度上仍然难以捉摸,部分原因是缺乏合适的技术来进行体内实时记录。
- 在这里,我们描述了 MTRIAOT,一种基于G蛋白偶联受体的绿色荧光 OT 传感器,具有大动态范围、合适的亲和力、对 OT 直系同源物的配体特异性、对下游信号传导的影响最小和长期荧光稳定性。
- 通过结合病毒基因传递和光纤光度法介导的荧光测量,我们展示了 MTRIAOT 用于实时检测活小鼠脑 OT 动态的效用。 MTRIAOT 介导的测量表明 OT 动力学的可变性取决于动物的行为背景和身体状况。 MTRIAOT 可能会在各种生理和病理过程中分析 OT 动力学。
16p11.2基因簇与CNS功能障碍
- 由反复拷贝数变异 (CNV) 引起的染色体 16p11.2 互惠基因组疾病涉及智力障碍、自闭症谱系障碍 (ASD) 和精神分裂症,但其相关机制尚不清楚。为了系统剖析分子效应,我们在含有同线 7qF3 区域的 CNV 以及细胞、转录、对 54 个等基因神经干细胞、诱导神经元和 CRISPR 工程 16p11.2 CNV 的脑类器官模型进行单细胞分析。
- 转录组范围内的差异表达基因在很大程度上是组织、细胞类型和剂量特异性的,尽管缺失和复制之间以及跨组织共享的影响比偶然预期的要多。在小脑(2,163 个差异表达基因)中观察到最广泛的影响,最大的富集与小鼠小脑和人类诱导的神经元中的突触通路有关。通路和共表达分析将能量和 RNA 代谢确定为 ASD 相关、功能丧失约束和脆弱 X 信使核糖核蛋白靶基因组的共享过程和富集。
- 有趣的是,相互的 16p11.2 剂量变化导致神经突和电生理特征的一致减少,并且类器官的单细胞分析显示兴奋性和抑制性 GABA 能神经元的比例相互改变。在我们的类器官分析中,神经元比率和基因表达的变化最直接地指向钙调蛋白 GABA 能抑制神经元和兴奋/抑制平衡作为破坏的目标,这可能有助于 16p11.2 携带者的神经发育和认知功能的变化。
- 总的来说,我们的数据表明基因组疾病涉及对多个贡献的生物过程的破坏,并且这种破坏具有特定于上下文的相对影响。
BRD4/ERα-RET-ERα超增强子正反馈回路
Super-enhancer-controlled positive feedback loop BRD4/ERα-RET-ERα promotes ERα-positive breast cancer. Nucleic Acids Res
- 雌激素和雌激素受体α (ERα) 诱导的基因转录与 ERα 阳性乳腺癌发生密切相关。 ERα占据的增强子,特别是超级增强子,已被认为在调节此类转录事件中起着至关重要的作用。然而,ERα 占据的超级增强子 (ERSE) 以及与 ERSE 相关的关键 ERα 诱导的靶基因仍有待充分表征。
- 在这里,我们定义了 ERα 阳性乳腺癌细胞系中 ERSE 的分布,并证明溴结构域蛋白 BRD4 是 ERSE 和同源 ERα 靶基因转录激活的主要调节因子。 RET 是酪氨酸蛋白激酶家族的成员,被鉴定为 BRD4 调节的 ERSE 的关键 ERα 靶基因,反过来,通过激活 RAS / 对 ERα 诱导的基因转录激活和恶性表型至关重要RAF/MEK2/ERK/p90RSK/ERα 磷酸化级联。
- BRD4 和 RET 抑制剂的联合治疗在体外和体内均表现出抑制 ERα 阳性乳腺癌的累加作用,与标准内分泌治疗他莫昔芬相当。此外,联合治疗使抗他莫昔芬的 ERα 阳性乳腺癌细胞系对他莫昔芬治疗重新敏感。
- 总之,我们的数据揭示了构成 BRD4/ERα-RET-ERα 的超增强子相关正反馈回路在 ERα 阳性乳腺癌中的关键作用,并表明该回路中的靶向成分将为治疗提供新的治疗途径临床上的ERα阳性乳腺癌。
lnRNA TERRA与端粒损伤修复
看上去蛮有趣的,可以了解一下
- 大量的人类癌症是端粒酶阴性的,并且通过称为端粒替代延长 (ALT) 的基于断裂诱导复制 (BIR) 的机制延长生理受损的端粒。
- 我们最近证明,抑制端粒长非编码 RNA TERRA 的转录可抑制端粒损伤和 ALT 特征,表明端粒转录是 ALT 活性的主要触发因素。
- 在这里,我们表明实验性增加的 TERRA 转录不仅如预期的那样增加了 ALT 特征,而且还通过需要核酸内切酶 Mus81 的途径导致端粒 DNA 的快速丢失。
- 我们的数据表明,如果控制不当,ALT 机制可能会危及端粒完整性,并指出 TERRA 转录是一种独特的多功能治疗靶点。
肿瘤ecDNA的进化动态
The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat Genet
了解一下研究方法
- 染色体外 DNA (ecDNA) 上的癌基因扩增是一种常见事件,它会推动肿瘤的侵袭性生长、耐药性和更短的生存期。目前,对非染色体癌基因遗传的影响——通过血统的随机身份——的影响知之甚少。同样不清楚的是 ecDNA 对体细胞变异和选择的影响。
- 在这里,我们将随机分离、无偏图像分析、基于 CRISPR 的 ecDNA 标记与活细胞成像和 CRISPR-C 的理论模型相结合,证明随机 ecDNA 遗传导致广泛的肿瘤内 ecDNA 拷贝数异质性和快速适应代谢应激和靶向治疗。观察到的 ecDNA 有益于宿主细胞的存活或生长,并且可以在单个细胞周期内发生变化。 ecDNA 遗传可以先验地预测含有 ecDNA 的癌症的一些侵袭性特征。 ecDNA 以一种通过染色体癌基因扩增无法实现的方式快速适应基因组的能力促进了这些特性。
- 这些结果显示了 ecDNA 的非染色体随机遗传模式如何导致癌症患者的不良预后。
机器学习/组学类
scRNA-seq数据库
HTCA: a database with an in-depth characterization of the single-cell human transcriptome. Nucleic Acids Res
- 单细胞 RNA 测序 (scRNA-seq) 是近几十年来最常用的单细胞组学之一。单细胞数据的指数级增长对于更能代表研究人群的大规模整合和深入探索具有巨大潜力。已努力整合已发表的数据,但仍缺乏广泛的表征。许多人专注于原始数据数据库的构建,而其他人则主要专注于基因表达查询。
- 在此,我们介绍了 HTCA (www.htcatlas.org),这是一个基于来自 3000 个 scRNA-seq 样本的 230 万个高质量细胞构建的交互式数据库,包含 19 个健康成人和匹配胎儿组织的深入表型谱。 HTCA 为成人和胎儿组织中跨细胞类型的基因特征、转录因子 (TF) 活动、TF 基序、受体-配体相互作用、丰富的基因本体 (GO) 术语等提供一站式交互式查询。
- 同时,HTCA 包括 16 种成人和胎儿组织的单细胞剪接变异谱、11 种成人和胎儿组织的空间转录组谱以及 27 种成人和胎儿组织的单细胞 ATAC 测序 (scATAC-seq) 谱。
- 此外,HTCA 提供在线分析工具来执行典型 scRNA-seq 分析中的主要步骤。
- 总之,HTCA 允许实时探索多组学成人和胎儿表型谱,并为灵活的 scRNA-seq 分析提供工具。
lncRNA免疫分型与ICI response及预后
免疫分型类研究。稍微关注一下。
- 长链非编码核糖核酸(RNA;lncRNA)与癌症免疫调节有关。然而,免疫细胞特异性 lncRNA 在胶质母细胞瘤 (GBM) 中的作用仍然很大程度上未知。
- 在这项研究中,构建了一个新的计算框架,通过使用六种机器学习方法综合分析纯化的免疫细胞、GBM 细胞系和大块 GBM 组织的转录组数据,筛选肿瘤浸润性免疫细胞相关 lncRNA (TIIClnc) 以开发 TIIClnc 特征。
- TIIClnc 特征可以区分 GBM 患者在四个独立数据集(包括湘雅内部数据集)中的生存结果,更重要的是,它显示出优于先前在胶质瘤中建立的 95 个特征的性能。
- TIIClnc 特征被揭示为免疫细胞浸润水平的指标,并预测免疫治疗的反应结果。 TIIClnc 签名与 CD8、PD-1 和 PD-L1 之间的正相关性在湘雅内部数据集中得到验证。
- 作为一种新证明的预测性生物标志物,TIIClnc 特征能够更精确地选择将从免疫治疗中受益的 GBM 人群,并应在不久的将来进行验证和应用。
协同药物组合预测网络SDCNet
Predicting cell line-specific synergistic drug combinations through a relational graph convolutional network with attention mechanism. Brief Bioinform
有一些AI的基础设计思路,可以参考下
- 由于组合的复杂性和 SDC 是细胞系特异性的事实,识别协同药物组合 (SDC) 是一项巨大的挑战。现有的计算方法要么没有考虑 SDC 的细胞系特异性,要么通过为每个细胞系独立构建模型而表现不佳。
- 在本文中,我们提出了一种名为 SDCNet 的新型编码器-解码器网络,用于预测细胞系特定的 SDC。SDCNet 在一个药物组合模型中学习不同细胞系的常见模式以及细胞系特异性特征。这是通过将不同细胞系的SDC图视为关系图,并构建关系图卷积网络(R-GCN)作为编码器来学习和融合药物对不同细胞系的深度表示来实现的。
- 设计了一种注意力机制,根据它们的相对重要性整合来自 R-GCN 不同层的药物特征,从而进一步增强表示学习。通过细胞系特定解码器中的部分参数共享来利用常见模式,该解码器不仅重建已知的 SDC,还预测每个细胞系的新 SDC。
- 对各种数据集的实验表明,SDCNet 优于最先进的方法,并且在推广到不同于训练细胞系的新细胞系时也很稳健。最后,案例研究再次证实了我们的方法在预测新的可靠细胞系特异性 SDC 方面的有效性。
预训练建模方法 SPRoBERTa用于蛋白质嵌入学习
SPRoBERTa: protein embedding learning with local fragment modeling. Brief Bioinform
- 很好地理解计算生物学中的蛋白质功能和结构有助于理解人类。面对有限的结构和功能注释的蛋白质,科学界采用了来自大量未标记蛋白质序列的自我监督预训练方法,用于蛋白质嵌入学习。然而,蛋白质通常由词汇量有限的单个氨基酸表示(例如 20 种类型的蛋白质),而没有考虑蛋白质序列中存在的强局部语义。
- 在这项工作中,我们提出了一种新颖的预训练建模方法 SPRoBERTa。我们首先提出了一个无监督的蛋白质标记器来学习具有局部片段模式的蛋白质表示。然后,引入了一种新的深度预训练模型框架来学习蛋白质嵌入。经过预训练,我们的方法可以很容易地针对不同的蛋白质任务进行微调,包括氨基酸水平预测任务(例如二级结构预测)、氨基酸对水平预测任务(例如接触预测)以及蛋白质水平预测任务(远程同源性预测,蛋白质功能预测)。
- 实验表明,我们的方法在所有任务中都取得了显着的改进,并且优于以前的方法。我们还为我们的蛋白质标记器和训练框架提供详细的消融实验(ablation studies)和分析。
混沌理论与深海动物潜水行为的结构发现
Strange attractor of a narwhal (Monodon monoceros). PLoS Comput Biol
听上去很新奇,可以了解一下
- 在海洋动物的持续潜水行为中检测结构具有挑战性,并且没有通用框架可用。我们使用混沌理论捕捉到了如此多样的结构。
- 通过对来自独角鲸的超长潜水记录(83 d)应用时间延迟嵌入,我们重建了状态空间肖像。使用混沌测量,我们检测到昼夜模式及其季节性调制、分类数据,并发现海冰外观如何改变时间预算。
- 在太阳正午有更多的近地表休息,但潜水更深,在黄昏和夜间潜水更激烈,但深度更浅(可能在鱿鱼之后);海冰外观减少休息。
- 引入的几何方法易于实施,并且可能有助于绘制和标记长期行为数据、识别个体动物和物种之间的差异以及检测扰动。
(~ ̄▽ ̄)~因果效应推断与致癌突变挖掘
Identification of key somatic oncogenic mutation based on a confounder-free causal inference model. PloS Comput Biol
蛮有趣的,有机会全文看一波!
- 异常细胞增殖和上皮间质转化 (EMT) 是诱导癌症发生和进展的重要事件。癌症研究的一个基本目标是开发一种有效的方法来检测能够驱动癌症的突变基因。尽管已经提出了几种计算方法来识别这些关键突变,但它们中的许多都关注基因突变与相关生物过程中功能变化之间的关联,而不是它们真正的因果关系。因果效应推断通过解决由于中性突变和未观察到的潜在变量引起的混杂偏差,提供了一种估计某种突变对癌症发生和进展的重要生物学过程的真实诱导效应的方法。
- 在这项研究中,整合基因组和转录组数据,我们构建了一个基于深度变分自动编码器的新型因果推理模型,以识别关键的致癌体细胞突变。应用于 10 种癌症类型,我们的方法通过减少观察到和未观察到的混杂偏差来量化基因突变对细胞增殖和 EMT 的因果影响。实验结果表明,突变频率较高的基因并不一定意味着它们在诱发癌症和促进癌症发展方面更有效。此外,我们的研究填补了使用机器学习进行因果推断以识别致癌突变的空白。
单细胞空间转录组分析方案EEL FISH
Scalable in situ single-cell profiling by electrophoretic capture of mRNA using EEL FISH. Nat Biotechnol
- 对转录组进行空间分析的方法主要是在分辨率和吞吐量之间进行权衡。在这里,我们开发了一种名为增强型电荧光原位杂交 (EEL FISH) 的方法,该方法可以在不影响空间分辨率的情况下快速处理大型组织样本。
- 通过将组织切片中的 RNA 电泳转移到捕获表面上,EEL 通过减少所需的成像量来加速数据采集,同时确保 RNA 分子直接向下移动到表面,从而保持单细胞分辨率。
- 我们将 EEL 应用于小鼠大脑的八个完整矢状切片,并测量多达 440 个基因的表达模式,以揭示复杂的组织组织。此外,EEL 可用于通过去除自发荧光脂褐质来研究具有挑战性的人体样本,从而使人类视觉皮层的空间转录组可视化。
- 我们为仪器控制、图像处理、数据分析和可视化提供完整的硬件规格、所有协议和完整的软件。
次等位基因频率检测方法SAIGE-GENE+
SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests. Nat Genet
可以了解一下
- 包括 UK Biobank (UKBB) 在内的多家生物样本库正在生成大规模测序数据。现有方法 SAIGE-GENE 在测试次要等位基因频率 (MAF) ≤ 1% 的变体时表现良好,但在限制 MAF ≤ 0.1% 或 0.01% 的变体时,在基于方差分量集的测试中观察到膨胀。
- 在这里,我们提出了 SAIGE-GENE+,它大大提高了 I 类错误控制和计算效率,以促进大规模数据中的罕见变体测试。我们进一步表明,结合多个 MAF 截止值和功能注释可以提高能力,从而揭示新的基因-表型关联。
- 在对 30 个定量性状和 141 个二元性状的 UKBB 全外显子组测序数据的分析中,SAIGE-GENE+ 确定了 551 个基因-表型关联。
豌豆品种ZW6参考基因组组装
可以了解一下植物的基因组从头组装和注释
- 完整而准确的参考基因组和注释为功能基因组学和作物育种提供了基础资源。在这里,我们报告了一个豌豆品种 ZW6 的从头组装和注释,其 contig N50 为 8.98 Mb,与现有的相比,其重叠群长度增加了 243 倍,复杂重复区域中序列的连续性和质量明显改善。
- 118 个栽培和野生豌豆的基因组多样性表明,Pisum abyssinicum 是不同于 P. fulvum 和 P. sativum 的一个独立物种。数量性状基因座分析揭示了两个已知的与茎长 (Le/le) 和种子形状 (R/r) 相关的孟德尔基因,以及孟德尔研究的一些豆荚形式的候选基因。
- 构建了116个豌豆种质的泛基因组,在P. abyssinicum和P. fulvum中优选的泛基因表现出明显的功能富集,表明它们在未来作为豌豆育种资源的潜在价值。
专家统一模型
An expert judgment model to predict early stages of the COVID-19 pandemic in the United States. Plos Comput BIol
我还以为是什么特别的思路,没想到是真*专家( ̄△ ̄;) 在PAD subtypes robustness的解释里可以引用此文的观点。
- 从 2020 年 2 月到 2020 年 5 月,传染病建模专家在一系列 13 项调查中提供了对新出现的 COVID-19 大流行趋势的定量预测和估计。大流行开始时有关现有传播模式的数据很少,但专家综合了他们可用的信息,以提供对当前和未来大流行状态的定量、基于判断的评估。
- 我们通过对他们的概率陈述进行同等加权平均,将专家预测聚合到一个“线性池”中。在很少有计算模型对大流行做出公开估计或预测的时候,专家判断提供了 (a) 与报告的 COVID-19 病例、住院和死亡相关的短期和长期大流行结果的可证伪预测,(b) 估计潜伏病毒传播,以及(c)在不同情况下对大流行轨迹的反事实评估。
- 聚合专家预测的线性池方法提供比任何单个专家更一致的准确预测,尽管线性池的预测准确性很少提供最准确的预测。
- 这项工作强调了专家线性池在未来新兴疫情早期灵活评估各种风险方面的重要性,特别是在可用数据尚不能支持数据驱动的计算建模的环境中。
致病基因识别新方法DGHNE
DGHNE: network enhancement-based method in identifying disease-causing genes through a heterogeneous biomedical network. Brief Bioinform
了解一下相关数据库“疾病基因关联数据库 DisGeNet”
- 致病基因的鉴定对于疾病病因学的机制理解和疾病预防和治疗的临床操作至关重要。然而,现有解决这个问题的方法在准确性和效率上都不够,需要具有更高识别能力的计算方法。
- 在这里,我们提出了一种称为 DGHNE 的新方法,通过网络增强支持的异构生物医学网络来识别致病基因。首先,通过疾病表型注释向量之间的余弦相似度得分构建疾病-疾病关联网络,并通过疾病-基因关联将疾病-疾病网络和基因-基因网络连接起来,构建新的异质生物医学网络。然后,通过使用基于高斯随机投影的网络嵌入,进一步增强了异构生物医学网络。最后,网络传播用于识别增强网络中的候选基因。
- 我们将 DGHNE 和其他五种方法一起应用到了最新的疾病基因关联数据库 DisGeNet 中。与所有其他方法相比,DGHNE 在全局 5 折交叉验证和预测新的疾病基因关联方面均显示了接收者操作特征曲线和精确召回曲线下的最高面积,以及最高的精确度和召回率。
- 我们进一步进行了 DGHNE 以确定帕金森病和糖尿病的候选致病基因,以及连接高血糖和糖尿病的基因。在所有情况下,预测的致病基因都富含与疾病相关的基因本体术语和京都基因百科全书和基因组途径,并且基因与疾病的关联得到了独立实验研究的高度证明。
多基因风险评分
ExPRSweb: An online repository with polygenic risk scores for common health-related exposures. Am J Hum Genet
可了解下这个评分的成分是什么
- 复杂性状受遗传风险因素、生活方式和环境变量(即所谓的暴露)的影响。一些暴露,例如吸烟或血脂水平,具有在全基因组关联研究中确定的常见遗传修饰因子。由于测量通常不可行,因此暴露多基因风险评分 (ExPRS) 提供了一种替代方法来研究暴露对各种表型的影响。
- 在这里,我们收集了 28 次曝光的公开汇总统计数据,并应用了四种常见的 PRS 方法在两个大型生物库中生成 ExPRS:密歇根基因组学计划和英国生物库。我们为 27 次暴露建立了 ExPRS,并证明了它们在全表型关联研究中的适用性以及作为常见慢性病的预测因子。特别是添加多个 ExPRS 显示,对于几种慢性病,与仅包括传统的、以疾病为中心的 PRS 的预测模型相比,有改进。
- 为了便于后续研究,我们通过名为 ExPRSweb 的在线存储库共享所有 ExPRS 结构和生成的结果。
炎症性肠病的血液biomarkers
From临床分子生物学研究所、基尔克里斯蒂安-阿尔布雷希特大学、德国基尔石勒苏益格-荷尔斯泰因大学医学中心、德国基尔石勒苏益格-荷尔斯泰因大学医学中心内科的Philip Rosenstiel团队
研究框架不错,可多加留意。
- 在 IBD 患者中使用肿瘤坏死因子 α (TNFα) 拮抗剂治疗的原发性无应答率高达 40%。缺少用于早期预测治疗成功的生物标志物。我们研究了接受 TNF 拮抗剂英夫利昔单抗治疗的 IBD 患者血液样本中基因表达和 DNA 甲基化的动态,并分析了治疗结果的预测潜力。
- 我们在两个接受首次英夫利昔单抗治疗的前瞻性 IBD 患者队列中进行了一项纵向的、基于血液的多组学研究(发现:14 名患者,复制:23 名患者)。在多达 7 个时间点收集样品(从基线到治疗诱导后 14 周)。分析了 RNA 测序和全基因组 DNA 甲基化数据,并将其与第 14 周的临床缓解相关联,作为主要终点。
- 我们发现两个队列之间没有一致的事前预测特征。非汇款组中纵向上调的转录本包括 TH2 和嗜酸性粒细胞相关基因,包括 ALOX15、FCER1A 和 OLIG2。网络构建确定了在基线和非缓解患者中一致表达但在缓解患者的早期时间点被破坏的转录模块。这些模块反映了干扰素信号传导、红细胞生成和血小板聚集等过程。 DNA 甲基化分析确定了缓解特异性的时间变化,这些变化与转录组信号部分重叠。机器学习方法识别出与第 2 周的 DNA 甲基化变化相关的差异表达基因的特征,作为第 14 周治疗结果的可靠预测因子,这在 20 名接受英夫利昔单抗治疗的 CD 患者的公开数据集中得到了验证。
- 综合多组学分析揭示了基因表达和 DNA甲基化的早期变化作为对抗TNF治疗有效反应的预测因子。缺乏此类特征可用于识别 IBD 患者在早期不太可能从 TNF 拮抗剂中受益。
快速算法YADA 3.0用于蛋白质谱反卷积
Increasing confidence in proteomic spectral deconvolution through mass defect. Bioinformatics
- 蛋白质组谱的可靠解卷积对于从头测序、交联质谱和处理嵌合质谱等多种应用至关重要。
- 一般来说,所有去卷积算法最终都可能报告与任何肽的化学式不兼容的质量峰。我们展示了如何通过考虑它们的质量缺陷来去除这些伪影。我们引入了 YADA 3.0,这是一种快速反卷积算法,可以去除具有不可接受的质量缺陷的峰。我们的方法对小于 10 kDa 的多肽有效,其本质可以很容易地整合到任何反卷积算法中。
- YADA 3.0 可在 http://patternlabforproteomics.org/yada3 上免费用于学术用途。
AlphaFold2与蛋白质序列比对
Highly significant improvement of protein sequence alignments with AlphaFold2. Bioinformatics
- 蛋白质序列比对对于结构、进化和功能分析至关重要,但它们的准确性通常受到序列相似性的限制,除非分子结构可用。因此,由 AlphaFold2 实现的以实验级准确度预测的蛋白质结构可能对序列分析产生重大影响。
- 在这里,我们发现根据 AlphaFold2 预测估计的多序列比对几乎与根据实验结构估计的比对一样准确,并且比基于序列的比对更接近结构参考。我们还表明,质量相对较低的 AlphaFold2 结构模型可用于获得高度准确的对齐。这些结果表明,除了结构建模之外,AlphaFold2 还编码了可用于序列分析的高阶依赖项。
- 可用性:所有数据、分析和结果都可在 Zenodo (https://doi.org/10.5281/zenodo.7031286) 上获得。代码和脚本已存放在 GitHub (https://github.com/cbcrg/msa-af2-nf) 和各种容器中 (https://cloud.sylabs.io/library/athbaltzis/af2/alphafold, https://hub.docker.com/r/athbaltzis/pred)。
单细胞多组学融合的深度学习框架
Integrated analysis of multimodal single-cell data with structural similarity. Nucleic Acids Res
看描述蛮有意思的,有机会可了解下
- 多模式单细胞测序技术从多层基因组读数中提供了有关细胞异质性的前所未有的信息。然而,在没有正确处理噪声的情况下对两种模态进行联合分析通常会导致一种模态与另一种模态过度拟合,并且聚类结果比普通单模态分析更差。如何有效地利用来自单细胞多组学的额外信息来描绘细胞状态并识别有意义的信号仍然是一项重大的计算挑战。
- 在这项工作中,我们提出了一个名为 SAILERX 的深度学习框架,用于对多模态单细胞数据进行高效、稳健和灵活的分析。 SAILERX 由具有不变表示学习的变分自编码器以纠正测序过程中的技术噪声,以及用于整合来自不同模态的信息的多模态数据对齐机制组成。
- SAILERX 不是通过将两种模态投射到共享的潜在空间来执行硬对齐,而是鼓励通过成对相似性测量的两种模态的局部结构相似。该策略对噪声的过度拟合更为稳健,有助于进行各种下游分析,如聚类、插补和标记基因检测。
- 此外,不变表示学习部分使 SAILERX 能够对多模态和单模态数据集进行综合分析,使其成为适用于更一般场景的可扩展工具。
基因组安全港识别
Genomics and epigenetics guided identification of tissue-specific genomic safe harbors. Genome Biol
蛮有意思的研究,有空看看全文和方法学
- 基因组安全港(Genomic safe harbors)是基因组中可以维持转基因表达而不破坏宿主细胞功能的区域。基因组安全港在提高基因组工程的效率和安全性方面发挥着越来越重要的作用。然而,已经确定了有限的安全港。
- 在这里,我们开发了一个框架,通过整合来自人类群体中自然发生的多态移动元素插入、表观基因组特征和 3D 染色质组织的信息来促进基因组安全港的搜索。通过将我们的框架应用于 1000 Genomes 项目和基因型组织表达 (GTEx) 项目中确定的多态性移动元素插入,我们确定了血细胞中的 19 个候选安全港和脑细胞中的 5 个候选安全港。
- 对于血液中的三个候选位点,我们证明了转基因的稳定表达,而不会破坏宿主红细胞中附近的基因。我们还开发了一个计算机程序,基因组学和表观遗传引导安全港映射器(GEG-SH 映射器),用于基于知识的组织特异性基因组安全港选择。
- 我们的研究提供了一个新的基于知识的框架来识别组织特异性基因组安全港。结合快速发展的基因组工程技术,我们的方法有可能在不久的将来提高基因和细胞疗法的整体安全性和效率。
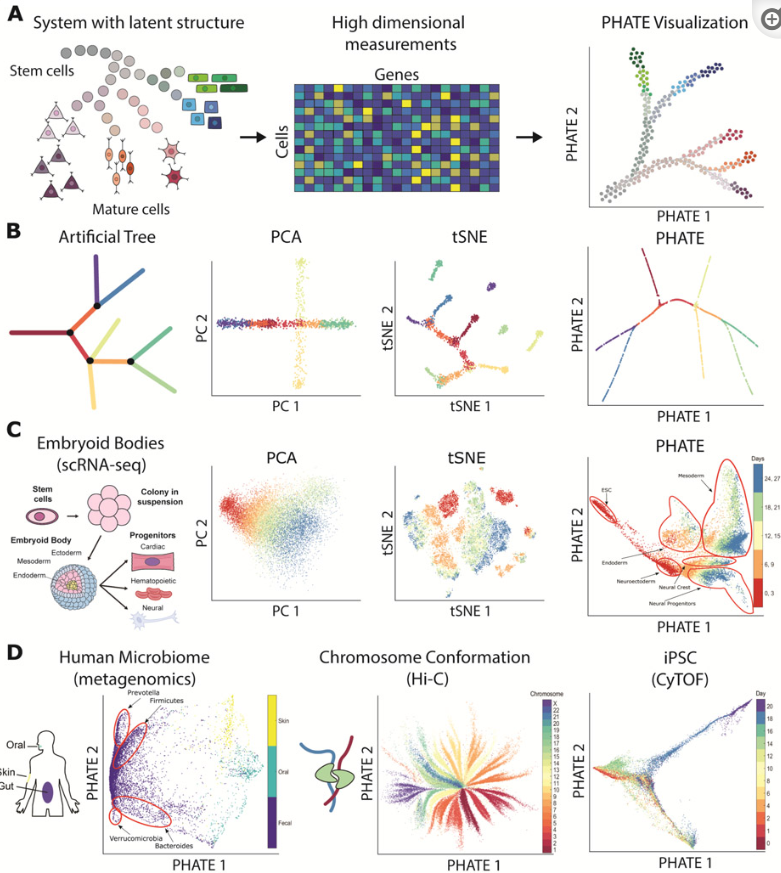
2019-PHATE与高维数据可视化
Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol
- 高通量技术创建的高维数据需要可视化工具,以直观的形式揭示数据结构和模式。
- 我们提出了 PHATE,这是一种可视化方法,它使用数据点之间的信息几何距离来捕获局部和全局非线性结构。
- 我们在各种人工和生物数据集上将 PHATE 与其他工具进行比较,发现它始终如一地保留了数据中的一系列模式,包括持续进展、分支和集群,优于其他工具。
- 我们定义了一个流形保存度量,我们称之为去噪嵌入流形保存 (DEMaP),并表明 PHATE 产生了与现有可视化方法相比在数量上更好地去噪的低维嵌入。对新生成的关于人类生殖层分化的单细胞 RNA 测序数据集的分析表明,PHATE 如何揭示对主要发育分支的独特生物学见解,包括识别三个以前未描述的亚群。
- 我们还表明,PHATE 适用于多种数据类型,包括大规模细胞术、单细胞 RNA 测序、Hi-C 和肠道微生物组数据。
角质形成细胞癌的表观遗传学亚型
Differentiation-related epigenomic changes define clinically distinct keratinocyte cancer subclasses. Mol Syst Biol. full.html
这个杂志多加关注,工作好像不太复杂
- 角质形成细胞癌 (KC) 是白皮肤人群中最常见的恶性肿瘤,给卫生系统带来了巨大的医疗和经济负担。 KC起源于表皮,主要包括基底细胞癌(BCC)和皮肤鳞状细胞癌(cSCC)。
- 在这里,我们结合单细胞多组学、转录组学和甲基组学来研究表皮分化过程中的表观基因组动力学。我们在未分化和分化的角质形成细胞之间确定了约 3,800 个差异可及区域,对应于与关键转录因子相关的调节区域。这些区域的 DNA 甲基化定义了具有表皮干细胞或角质形成细胞样特征的 AK/cSCC 亚型。
- 使用细胞类型去卷积工具和整合大量和单细胞甲基化组,我们证明这些亚类与不同的细胞来源一致。对亚类表型特征的进一步表征和对其他未分层 KC 实体的研究揭示了亚类的不同临床特征,将侵袭性和转移性 KC 病例与未分化的细胞来源联系起来。
- 我们的研究对人类角质形成细胞分化的表观基因组动力学进行了全面的表征,并揭示了 KC 起源细胞与其预后之间的新联系。
细胞重编程的小分子化合物组合识别
Small compound-based direct cell conversion with combinatorial optimization of pathway regulations. Bioinformatics
the problem of combinatorial explosion 组合爆炸 的描述非常形象
研究的领域也十份有趣。但是这个代码托管就有点随意了。
- 直接细胞转化或直接重编程 (DR),是一种创新技术,可将源细胞直接转化为靶细胞,而无需绕过诱导的多能干细胞。使用小分子化合物(例如药物)进行 DR 可以帮助避免基因转染引起的致癌风险;然而,由于组合爆炸,通过实验识别小化合物仍然具有挑战性。
- 结果:在本文中,我们提出了一种新的计算方法 COMPRENDRE(用于直接重编程的途径调节的组合优化),以阐明基于小化合物的 DR 的机制并预测用于 DR 的小化合物的新组合。我们估计了 DR 诱导小化合物的潜在目标蛋白,并确定了一组涉及 DR 的目标途径。我们确定了多种 DR 相关途径,这些途径以前没有报道过从成纤维细胞中诱导神经元或心肌细胞。为了克服组合爆炸的问题,我们开发了一种模拟退火算法的变体,以确定可以调节 DR 相关途径的最佳化合物集。因此,所提出的方法能够用更少的化合物预测新的 DR 诱导候选组合,并成功地复制经过实验验证的化合物,诱导从成纤维细胞直接转化为神经元或心肌细胞。所提出的方法有望用于再生医学的实际应用。
- 可用性和实施:支持当前研究的代码可在 http://labo.bio.kyutech.ac.jp/~yamani/comprendre 获得。
DNA结合预测的深度学习pipeline
DeepZF: improved DNA-binding prediction of C2H2-zinc-finger proteins by deep transfer learning. Bioinformatics
- Cys2His2 锌指 (C2H2-ZF) 蛋白是人类转录因子中最大的一类,因此在基因调控和细胞功能中发挥着核心作用。 C2H2-ZF 蛋白的特征在于包含多个 ZF 的 DNA 结合结构域。 ZF 的一个子集结合不同的 DNA 三联体。尽管它们发挥着核心作用,但对于它们的哪些 ZF 是结合的,以及 DNA 结合偏好如何在每个 ZF 的氨基酸序列中编码,我们知之甚少。
- 我们提出了 DeepZF,这是一个基于深度学习的管道,用于预测结合 ZF 及其 DNA 结合偏好,仅给出 C2H2-ZF 蛋白的氨基酸序列。据我们所知,我们编译了第一个结合和非结合 ZF 的体内数据集,用于训练第一个 ZF 结合分类器。我们的分类器基于一种新型蛋白质转换器,平均 AUROC 为 0.71。此外,我们利用体内和体外数据集通过迁移学习来学习 ZF-DNA 结合的识别码。我们新开发的模型是第一个将深度学习用于该任务的模型,在三个 DNA 结合位置中的每一个上都实现了大于 0.94 的平均 Pearson 相关性。
- 总之,DeepZF 在 C2H2-ZF 蛋白 DNA 结合偏好预测任务中的表现优于现有方法:它在基序相似性方面实现了 0.42 的平均 Pearson 相关性,而现有方法实现的平均相关性小于 0.1。
- 通过应用已建立的可解释性技术,我们表明 DeepZF 推断了生物学相关的结合原理,例如氨基酸残基位置对 ZF DNA 结合潜力的影响。
- 可通过 https://github.com/OrensteinLab/DeepZF 获得 DeepZF 代码、模型和结果。
投影细胞测量的新方法Synmatch
Linking cells across single-cell modalities by synergistic matching of neighborhood structure. Bioinformatics
- 有多种实验方法可用于表征复杂生物样品中单细胞的不同特性。然而,由于这些测量技术通常具有破坏性,因此研究人员通常会收到来自不相交的细胞亚群的互补测量,从而提供细胞生物过程的碎片化视图。这就需要能够集成不相交的多组学数据的计算工具。因为不同的测量通常不共享任何特征,所以这个问题需要以无监督的方式进行集成。最近,已经提出了几种方法,将细胞测量投影到一个共同的潜在空间中,并尝试对齐相应的低维流形。
- 在这项研究中,我们提出了一种方法 Synmatch,该方法通过利用每个模态中的邻域结构信息来产生模态之间的单元格的直接匹配。同步匹配依赖于直觉,即在一个测量空间中靠近的单元在另一个测量空间中也应该靠近。这使我们能够将匹配问题表述为邻域结构上的约束超模优化问题,可以有效地求解。我们表明,我们的方法成功地匹配了小型真实多组学数据集中的细胞,并且与最近发布的最先进的方法相比表现良好。此外,我们证明了 Synmatch 能够扩展到包含数千个细胞的大型数据集。
- 本手稿中使用的 Synmatch 代码和数据可在 https://github.com/Noble-Lab/synmatch 获得。
pseudo-bulk RNA-seq 数据模拟
SimBu: bias-aware simulation of bulk RNA-seq data with variable cell-type composition. Bioinformatics
这个研究很有趣
- 由于复杂组织通常由各种细胞类型组成,因此已开发出反卷积工具,以从大量 RNA 测序 (RNA-seq) 数据中计算推断其细胞组成。要全面评估反卷积性能,金标准数据集是必不可少的。流式细胞术或免疫组织化学等金标准实验技术是资源密集型的,不能系统地应用于通过高通量转录组学分析的众多细胞类型和组织。通过以预定义的比例聚合单细胞 RNA-seq 表达谱生成的“伪大量”数据的模拟提供了一种可扩展且具有成本效益的替代方案。这使得创建计算机金标准成为可能,该标准允许对实验设置中无法想象的细胞类型分数进行细粒度控制。然而,目前还没有用于生成pseudo-bulk RNA-seq 数据的模拟软件。
- 我们开发了 SimBu,一个能够基于各种模拟场景模拟伪散装样本的 R 包,旨在测试反卷积方法的特定特征。 SimBu 的一个独特功能是使用实验衍生或数据驱动的比例因子对细胞类型特异性 mRNA 偏差进行建模。在这里,我们展示了 SimBu 可以生成逼真的伪批量数据,概括真实 RNA-seq 数据的生物学和统计特征。最后,我们说明了 mRNA 偏差对反卷积工具评估的影响,并为选择合适的估计 mRNA 含量的方法提供了建议。 SimBu 是一种用户友好且灵活的工具,用于模拟真实的伪批量 RNA-seq 数据集,作为评估细胞类型反卷积方法的计算机黄金标准。
- SimBu 在 https://github.com/omnideconv/SimBu 作为 GPL-3 许可下的 R 包免费提供。
GWAS线性混合模型优化算法
Efficient permutation-based genome-wide association studies for normal and skewed phenotypic distributions. Bioinformatics
- 全基因组关联研究 (GWAS) 是研究复杂基因型和表型关系结构的不可或缺的工具。线性混合模型 (LMM) 通常用于检测遗传标记和感兴趣的性状之间的关联,同时允许考虑种群结构和隐秘的相关性。 LMM 的假设包括残差的正态分布以及遗传标记是独立且同分布的——这两个假设在实际数据中经常被违反。基于排列的方法可以帮助克服其中的一些限制,并为发现真正的关联提供更现实的阈值。尽管如此,在实践中,由于计算复杂度高,它们很少被实现。
- 我们提出了 permGWAS,这是一种基于4D张量的有效 LMM 重构,可以提供基于排列的显着性阈值。我们表明,我们的方法在运行时间方面优于当前最先进的 LMM,并且与常用的 Bonferroni 阈值相比,基于排列的阈值对偏斜表型的错误发现率更低。此外,使用 permGWAS,我们在不到 8 天的时间内在单个 GPU 上重新分析了 500 多个拟南芥表型,每个表型有 100 个排列。我们的重新分析表明,应用基于排列的阈值可以改进和完善对 GWAS 结果的解释。
- permGWAS 是开源的,可在 GitHub 上公开下载:https://github.com/grimmlab/permGWAS。
药物重定位的深度学习框架
A geometric deep learning framework for drug repositioning over heterogeneous information networks. Brief Bioinform
- 药物重新定位(DR)是一种很有前途的策略,可以利用人工智能技术发现已批准药物的新指标,从而改善传统药物的发现和开发。然而,大多数 DR 计算方法都没有考虑到生物医学网络数据的非欧几里德性质。
- 为了克服这个问题,提出了一种深度学习框架,即 DDAGDL,通过在异构信息网络 (HIN) 上使用几何深度学习 (GDL) 来预测药物-药物关联 (DDA)。 DDAGDL 将复杂的生物信息融入 HIN 的拓扑结构中,通过注意力机制有效地学习药物和疾病的平滑表示。
- 实验结果表明,与最先进的 DR 方法相比,DDAGDL 在 10 倍交叉验证下的三个真实数据集上在几个评估指标方面的优越性能。
- 我们的案例研究和分子对接实验表明,DDAGDL 是一种很有前途的 DR 工具,它获得了利用几何先验知识提高功效的新见解。
B细胞受体和抗体特征的表征工具AIRRscape
AIRRscape: An interactive tool for exploring B-cell receptor repertoires and antibody responses. PloS Comput Biol
Shiny可以写到GSClassifier的TODO list里去
- B 细胞抗体库的测序覆盖范围和深度不断增加,从而鉴定出大量免疫球蛋白重链和轻链。然而,这些自适应免疫受体库测序 (AIRR-seq) 数据集的大小和复杂性使得进行探索性分析变得困难。
- 为了帮助进行数据探索,我们开发了 AIRRscape,这是一个基于 R Shiny 的交互式 Web 浏览器应用程序,它可以通过多个库之间的比较来发现 B 细胞受体 (BCR) 和抗体特征。
- 使用 AIRR-seq 数据作为输入,AIRRscape 首先将所有曲目聚合和分类到种系 V 基因、种系 J 基因和 CDR3 长度的交互式和可探索箱中,提供整个曲目的高级视图。可以快速识别和选择感兴趣的曲目子集,然后可以生成 CDR3 基序的网络拓扑以供进一步探索。
- 在这里,我们使用患者 BCR 库和已发表的单克隆抗体序列展示了 AIRRscape,以研究对三种病毒病原体的体液免疫模式:SARS-CoV-2、HIV-1 和 DENV(登革热病毒)。 AIRRscape 揭示了所有三种病原体数据集中的收敛抗体序列,尽管 HIV-1 抗体数据集显示有限的收敛和特殊反应。
- 我们已将 AIRRscape 作为基于 Web 的 Shiny 应用程序以及 GitHub 上的代码提供,以鼓励免疫信息学家、病毒学家、免疫学家、疫苗开发人员和其他有兴趣探索和比较多种免疫受体的科学家开放开发和使用曲目。
DNA甲基化与组蛋白修饰
DNA methylation underpins the epigenomic landscape regulating genome transcription in Arabidopsis. Genome Biol
- 由于缺乏无 DNA 甲基化突变体,确定 DNA 甲基化对表观遗传景观和高等生物功能的影响具有挑战性。
- 在这里,对最近产生的完全没有 DNA 甲基化的拟南芥突变体的分析表明,DNA 甲基化是组蛋白修饰的全基因组景观的基础。 DNA 甲基化的完全丧失导致组蛋白修饰格局的剧变,包括 H3K9me2 的完全丧失以及活性和 H3K27me3 组蛋白标记的广泛重新分布,这主要是由于 DNA 甲基化在启动 H3K9me2 沉积中的作用并排除了活性标记和抑制标记 H3K27me3; CG 和非 CG 甲基化可以在某些基因组区域独立发挥作用,而在许多其他区域协同作用。
- 失去所有 DNA 甲基化后的转录重编程与所检查的组蛋白修饰的广泛重新分布或转换相关。在无 DNA 甲基化突变体中保留或获得的组蛋白修饰用作 DNA 甲基化非依赖性转录调节信号:活性标记促进基因组转录,而抑制性标记 H3K27me3 弥补了多个转座子家族中 DNA 高甲基化/H3K9me2 的缺乏。
- 我们的研究结果表明,完整的 DNA 甲基化组构成了拟南芥表观基因组景观的支架,对于受控的基因组转录和最终的正常生长和发育至关重要。
转录因子预测
- 了解蛋白质如何与 DNA 相互作用对于理解基因调控至关重要。尽管已经确定了数千种转录因子 (TF) 的 DNA 结合特异性,但构成其结构界面的特定氨基酸碱基相互作用在很大程度上是未知的。这种分辨率的缺乏阻碍了利用这些数据来预测未表征的 TF 或疾病中突变的 TF 的特异性的尝试。
- 在这里,我们通过蛋白质-DNA 结构界面 (rCLAMPS) 的自动映射引入识别码学习,这是一种概率方法,它使用来自同一结构家族的 TF 的 DNA 结合特异性来同时推断哪些核苷酸位置被特定氨基酸接触。 TF 以及将每个碱基接触氨基酸与其接触的 DNA 位置的核苷酸偏好相关联的识别代码。我们将 rCLAMPS 应用于同源域,即后生动物中的第二大 TF 家族,并表明它学习了一种高效的识别代码,可以预测 TF 的从头 DNA 结合特异性。
- 此外,我们表明推断的氨基酸-核苷酸接触揭示了个体结合位点位置的核苷酸偏好是否以及如何被 TF 内的突变改变。我们的方法是朝着从 DNA 结合特异性的大量纲要中自动揭示蛋白质-DNA 特异性的决定因素并推断疾病中突变的 TF 功能改变的重要一步。
胶质瘤中的3种恶性间充质状态
Elucidating the diversity of malignant mesenchymal states in glioblastoma by integrative analysis. Genome Med
- 多项胶质母细胞瘤研究描述了间充质 (MES) 状态,每项研究都通过不同的基因组定义 MES 程序,并强调不同的功能关联,包括免疫激活和抑制。这些变量描述使我们对 MES 状态及其含义的理解复杂化。
- 在这里,我们假设存在一系列神经胶质瘤 MES 状态,可能反映了可以诱导 MES 程序的不同先前状态,和/或在这些细胞中诱导 MES 状态的不同机制。
- 我们整合了多个已发表的单细胞和批量 RNA 测序数据集和 MES 特征,以定义跨研究重复出现的核心 MES 程序,以及在 MES 细胞之间变化的多个功能特异性 MES 特征。然后,我们检查了这些特征的共现及其与遗传和微环境特征的关联。
- 基于 MES 特征的共现,我们发现了 MES 状态的三种主要变体:缺氧相关 (MES-Hyp)、星形胶质细胞相关 (MES-Ast) 和中间状态。值得注意的是,MES 状态与遗传和微环境特征存在差异。 MES-Hyp 优先与 NF1 缺失、整体巨噬细胞丰度、高巨噬细胞/小胶质细胞比率和 M2 相关巨噬细胞相关,这与之前将 MES 与免疫抑制相关的研究一致。相比之下,MES-Ast 与 T 细胞丰度和细胞毒性相关,这与通过 MHC-I/II 表达的免疫激活一致。
- 胶质母细胞瘤中存在多种 MES 状态。这些状态共享核心基因的一个子集,但主要区别在于它们与缺氧与星形细胞表达程序的关联,以及与免疫抑制与激活的关联。
非裔的TNBC的RNA测序结果
老实说,最近这种关注少数族裔的研究很多,特别是非裔,而且还发表在特别好的杂志下(以相对普通的研究工作)。关注一下亚裔相关研究。
- 撒哈拉以南非洲裔女性的三阴性乳腺癌 (TNBC) 发病率和 TNBC 特异性死亡率高得不成比例。人群比较研究显示 TNBC 生物学存在种族差异,包括非裔美国人 (AA) 中基底样和四阴性亚型的患病率较高。然而,之前的调查主要依赖于美国 (US) 人口的自我报告种族 (SRR)。由于异源性遗传混合物和社会决定因素的生物学后果,非洲血统与 TNBC 生物学的真正关联尚不清楚。
- 为了解决这个问题,我们使用 TNBC 对 AA、西非和东非的国际队列进行了 RNAseq。在这个非洲人丰富的队列中使用全面的遗传血统估计,我们发现了 613 个与非洲血统相关的基因和 2000 多个与区域非洲血统相关的基因的表达。
- 非洲相关基因的一个子集也显示出正常乳腺组织的差异。肿瘤细胞组成的通路富集和去卷积揭示了非洲裔患者的肿瘤相关免疫学特征是不同的。
免疫组化的多任务AI框架DeepLIIF
Deep Learning-Inferred Multiplex ImmunoFluorescence for Immunohistochemical Image Quantification. Nat Mach Intell
来自美国纽约斯隆凯特琳纪念癌症中心医学物理系的Saad Nadeem团队
很朴实的研究,有时间看看全文。此研究可能会讨论IHC切片特征提取准备工作中的困难。
- 通过组织的常规免疫组织化学 (IHC) 染色评估的报告生物标志物广泛用于诊断病理学实验室的患者护理。迄今为止,临床报告主要是定性或半定量的。
- 通过创建称为 DeepLIIF 的多任务深度学习框架,我们提出了一种单步解决方案来染色反卷积/分离、细胞分割和定量单细胞 IHC 评分。利用共同注册的 IHC 和同一载玻片的多重免疫荧光 (mpIF) 染色的独特从头数据集,我们将低成本和流行的 IHC 载玻片分割并转换为更昂贵但信息量更大的 mpIF 图像,同时提供必要的基础叠加明场 IHC 通道的真相。此外,引入了具有高 (>95%) 细胞覆盖率的新核包膜染色剂 LAP2beta,以改善 IHC 载玻片上的细胞描绘/分割和蛋白质表达量化。通过同时将输入 IHC 图像转换为干净/分离的 mpIF 通道并执行细胞分割/分类,
- 我们表明,我们在干净的 IHC Ki67 数据上训练的模型可以推广到更多噪声和伪影较多的图像以及其他核和非核标记如 CD3、CD8、BCL2、BCL6、MYC、MUM1、CD10 和 TP53。我们在公开可用的基准数据集以及病理学家的半定量评分上彻底评估了我们的方法。代码、预训练模型以及易于运行的容器化 docker 文件以及 Google CoLab 项目可在 https://github.com/nadeemlab/deepliif 获得。
无监督聚类方法MODEC
MODEC: an unsupervised clustering method integrating omics data for identifying cancer subtypes. Brief Bioinform
重点关注!
- 癌症亚型的鉴定可以帮助研究人员了解隐藏的基因组机制,提高诊断准确性并改进临床治疗。随着高通量技术的发展,研究人员可以从多个来源访问大量数据。由于多组学和临床数据的高维和复杂性,需要对多组学数据的整合进行研究,而为此目的开发有效工具仍然是研究人员面临的挑战。
- 在这项工作中,我们提出了一种完全无监督的聚类方法,无需利用任何先验知识(MODEC)。我们使用多种优化和深度学习技术来整合多组学数据,以识别癌症亚型和分析重要的临床变量。
- 由于基因级数据集存在非线性,我们使用流形优化方法从原始组学数据中提取基本信息,以获得低维潜在子空间。然后,MODEC 使用基于深度学习的聚类模块迭代地定义聚类质心,并通过最小化 Kullback-Leibler 散度损失为每个样本分配聚类标签。
- MODEC 被应用于癌症基因组图谱数据库中的六个公共癌症数据集,并在亚型结果的准确性和可靠性方面优于八种竞争方法。
- MODEC 在识别生存模式和重要临床特征方面极具竞争力,可以帮助医生监测疾病进展并提供更合适的治疗策略。
基于孟德尔随机化的多组学融合框架
Integrating multi-omics summary data using a Mendelian randomization framework . Brief Bioinform
- 孟德尔随机化(Mendelian randomization)是一种通用工具,可使用遗传变异作为工具变量来识别组学生物标志物与疾病结果之间可能的因果关系。一个关键主题是基因的优先级,其组学读数可通过分析 GWAS 和 QTL 汇总数据用作疾病结果的预测因子。然而,在探索注释到同一感兴趣基因的多个组学生物标志物的影响方面,缺乏最佳实践的研究。
- 为了弥合这一差距,我们提出了强大的组合测试,它整合了多个相关的 $P$ 值,而不假设暴露之间的依赖结构。
- 我们广泛的模拟实验证明了我们提出的方法与适应我们感兴趣的环境的现有方法相比的优越性。
- 多组学阿尔茨海默病数据集分析的热门话题包括基因 ABCA7 和 ATP1B1。
mRNA表达节律的进化起源
Rhythmicity is linked to expression cost at the protein level but to expression precision at the mRNA level. PLoS Comput Biol
- 许多基因具有夜间表达节律,即 24 小时周期性变化,在 mRNA 或蛋白质水平或两者兼有,大多数节律基因是组织特异性的。在这里,我们调查和讨论基因表达节律的进化起源。
- 我们的结果表明,蛋白质表达的节律性可能受到选择的青睐,以最大限度地降低成本。细菌、植物和动物的趋势是一致的,并且也得到了小鼠组织特异性模式的支持。
- 与蛋白质水平不同,成本无法解释 RNA 水平的节律。我们建议相反,它允许定期减少表达噪音。噪音控制在老鼠身上得到了最有力的支持,在其他物种中的证据有限。我们还发现,在更强的纯化选择下,基因在 mRNA 水平有节奏地表达,我们认为这是因为它们是噪声敏感基因。
- 最后,节律表达的适应性作用得到了高度表达但组织特异性的节律基因的支持。这为夜间节律通常是组织特异性的观察提供了很好的进化解释。
综述类
癌症治疗中的设备管理
Prevention of device-related infections in patients with cancer: Current practice and future horizons. CA Cancer J Clin
蛮有意思的,值得一看
- 在过去的几年中,癌症管理的多方面进展导致生存率显着提高。在患者的整个肿瘤学旅程中,他们可能会收到一种或多种植入式设备,用于输液和药物管理,以及管理与癌症治疗相关的各种合并症和并发症。与这些设备相关的感染频繁且复杂,通常需要移除设备,增加医疗保健成本,对生活质量产生负面影响,并使肿瘤护理复杂化,通常会导致进一步挽救生命的癌症治疗延迟。
- 在此,作者全面回顾了多项基于证据的建议,以及预防各种设备相关感染的最佳实践、专家意见和新方法。作者提出了许多预防这些感染的一般原则,然后系统地提出了与设备相关的具体建议。监管实体、行业、专业医学协会、医院和针对感染控制的干预措施以及初级保健和咨询医疗保健提供者之间的持续参与和有意义的合作对于持续降低这些可预防感染的发生率都至关重要。
肿瘤PDX模型
Towards precision oncology with patient-derived xenografts. Nat Rev Clin Oncol
- 在治疗的选择压力下,肿瘤动态地进化出多种适应性机制,使得对基因组改变的静态询问不足以指导治疗决策。临床研究无法评估肿瘤中的各种调节回路如何随着时间和空间受到治疗性损伤的影响。同样,在患者身上测试由复合和不断变化的分子信息提供的不同精准肿瘤学方法也很难实现。因此,需要结合人类癌症生物学和遗传学的临床前模型,促进复杂变量的分析并实现足够的人口吞吐量,以查明随机分布的响应预测因子。
- 源自患者的异种移植 (PDX) 模型是动态实体,其中可以通过小鼠中的连续传播来监测癌症的演变。 PDX 模型还可以概括患者间的多样性,从而能够识别分子定义的肿瘤亚组的反应生物标志物和治疗靶点。
- 在这篇综述中,我们讨论了过去十年使用 PDX 模型进行精准肿瘤学的例子,从转化研究到药物发现。我们详细阐述了 PDX 模型中的临床前观察结果如何以及在多大程度上证实和/或预期在患者中的发现。最后,我们说明了新兴的方法论努力,这些努力可以通过提高预测准确性或提高其多功能性来扩大 PDX 模型的应用。
人类端粒相关疾病
Genetics of human telomere biology disorders. Nat Rev Genet
- 端粒是位于线性染色体末端的特殊核蛋白结构,可防止 DNA 损伤反应和修复途径的激活。许多因素定位于端粒以调节其长度、结构和功能,以避免复制性衰老或基因组不稳定和细胞死亡。
- 在人类中,其中几个因素的孟德尔缺陷可导致端粒异常短或功能失调,从而导致一组罕见的异质性过早衰老疾病,称为端粒病、短端粒综合征或端粒生物学疾病 (TBD)。
- 在这里,我们回顾了迄今为止确定的导致 TBD 的基因,并描述了它们与端粒生物学相关的主要功能。我们介绍了 TBD 的分子方面,包括遗传预期、表型、不完全外显率和体细胞遗传拯救,这些是这些疾病复杂性的基础。
- 我们还讨论了 TBD 的表型和遗传特征对与人类端粒生物学、衰老和癌症相关的基本方面以及诊断、治疗和临床方法的影响。
RNA-Seq数据处理
Normalizing cancer RNA-seq data for library size, tumor purity and batch effects. Nat Biotechnol
尚无摘要。但要持续关注!!
精神遗传学
Ten challenges for clinical translation in psychiatric genetics. Nat Genet
- 全基因组关联研究已经确定了数百种潜在的精神疾病的强大遗传关联,并为疾病的发作和进展提供了重要的生物学见解。人们乐观地认为,基因发现将为精准精神病学铺平道路,促进开发更有效的治疗方法,并确定这些治疗应针对的患者群体。然而,在将基因发现转化为临床之前,必须解决几个挑战。
- 在本期展望中,我们强调了精神遗传学领域面临的十大挑战,重点是对遗传风险因素的稳健和普遍检测,改进精神病理学的定义和评估,以及实现更好的临床指标。我们讨论了该领域的最新进展,这些进展将提高遗传数据的解释和预测能力,并最终有助于改善精神疾病患者的管理和治疗。
药物开发数据集
A review of biomedical datasets relating to drug discovery: a knowledge graph perspective. Brief Bioinform
这个综述看上去不错,值得了解;proximity metric
- 药物发现和开发是一个复杂且成本高昂的过程。正在研究机器学习方法,以帮助提高药物发现管道多个阶段的有效性和速度。其中,那些使用知识图谱 (KG) 的人在许多任务中都有希望,包括药物再利用、药物毒性预测和目标基因疾病优先级排序。在药物发现知识库中,包括基因、疾病和药物在内的关键要素被表示为实体,而它们之间的关系则表示相互作用。但是,要构建高质量的 KG,需要合适的数据。
- 在这篇综述中,我们详细介绍了适用于构建以药物发现为重点的 KG 的公开可用资源。我们的目标是帮助指导有兴趣将新技术应用于药物发现领域但可能不熟悉相关数据源的机器学习和 KG 从业者。数据集是通过严格的标准选择的,根据其中包含的主要信息类型进行分类,并根据可以提取哪些信息来构建 KG 进行考虑。
- 然后,我们对现有的公共药物发现 KG 进行比较分析,并对文献中选定的激励案例研究进行评估。此外,我们提出了与该领域及其数据集相关的众多独特挑战和问题,同时还强调了未来的主要研究方向。我们希望这篇综述能激发 KG 用于解决药物发现领域的关键问题和新出现的问题。
生物医学数据处理
Linking research of biomedical datasets. Brief Bioinform
可以了解一下作者的中心思想
- 生物医学数据预处理和高效计算与用于拟合数据的统计方法一样重要;数据处理需要考虑应用场景、数据获取和个人权益。
- 我们根据全流程处理机制的多样性、连贯性、共享性、可审计性和生态性,回顾了集成研究的共同原则、知识和方法。首先,神经形态和原生算法集成了不同的数据集,提供线性可扩展性和高度可视化。其次,总结了节点和协调器平台上从原始到神经形态的不同预处理、分析和事务方法的选择机制。三是节点、网络、云、边、群、图相结合,构建队列整合研究和临床诊疗的生态系统。
- 展望未来,同时结合深度计算、海量数据存储和大规模并行通信至关重要。
免疫治疗biomarkers
Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther
很重要的综述,找全文看。
- 癌症是高度复杂的疾病,其特征不仅在于恶性细胞的过度生长,还在于免疫反应的改变。免疫系统的抑制和重编程在肿瘤的发生和发展中起关键作用。免疫疗法旨在重新激活抗肿瘤免疫细胞并克服肿瘤的免疫逃逸机制。
- 以免疫检查点阻断和过继细胞转移为代表,肿瘤免疫疗法在临床上取得了巨大成功,能够诱导一些对所有其他治疗无效的肿瘤长期消退。其中,以PD-1/PD-L1抑制剂(nivolumab)和CTLA-4抑制剂(ipilimumab)为代表的免疫检查点阻断疗法在多种恶性肿瘤的治疗中显示出令人鼓舞的疗效。
- 此外,随着CAR-T、CAR-M等新型免疫治疗方法的问世,免疫治疗进入了一个新时代。目前,有证据表明多种免疫治疗方法的结合可能是提高治疗效果的一种方法。然而,肿瘤免疫治疗的整体临床反应率仍有待提高,这需要开发新的治疗设计以及发现可以指导这些药物处方的生物标志物。
- 从过去临床和基础研究的成功和失败中吸取教训对于未来研究的合理设计至关重要。在本文中,我们描述了操纵免疫系统对抗癌症的努力,并讨论了可用于促进抗肿瘤免疫反应的不同靶标和细胞类型。
靶向淋巴结的肿瘤纳米疫苗
Lymph node-targeting nanovaccines for cancer immunotherapy. J Control Release
- 肿瘤疫苗、嵌合抗原受体T细胞和免疫检查点阻断等癌症免疫疗法引起了极大的关注。其中,肿瘤疫苗通过向抗原呈递细胞(APC)递送抗原和佐剂来引发免疫反应,从而增强抗肿瘤免疫治疗。尽管肿瘤疫苗在肿瘤免疫治疗方面取得了相当大的成就,但有效地递送肿瘤疫苗以激活淋巴结 (LN) 中的树突状细胞 (DC) 仍然具有挑战性。
- 基于生物医学纳米技术的纳米疫苗的合理设计已成为提高癌症免疫治疗效果的最有希望的策略之一。近年来,人们在开发各种基于纳米载体的LNs靶向肿瘤纳米疫苗方面做出了巨大努力。
- 鉴于该领域的快速发展,我们在此旨在总结用于癌症免疫治疗的 LNs 靶向纳米疫苗的最新进展,特别关注为 LNs 靶向递送肿瘤疫苗而开发的各种纳米载体,包括基于脂质的纳米颗粒、聚合物纳米载体、无机纳米载体和仿生纳米系统。此外,还提供了基于纳米疫苗的联合癌症免疫疗法的最新趋势。最后,聚焦LNs靶向纳米疫苗在临床转化和应用中的合理性、优势和挑战。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍
