本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- ZBP1 和 cGAS 协同感知线粒体 DNA 促…
- 肌动蛋白丝的自由端和加帽端的结构
- 油菜素类固醇通过细胞壁和组织力学协调植物细胞层相互…
- 紫外线辐射塑造皮肤树突状细胞白血病转化
- Perforin-2 是交叉提呈树突状细胞的内吞逃…
前言
本文是前沿快讯的第31期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。从第18期开始,“肿瘤转移类”、“肿瘤代谢类”等将不再更新,而是合并至其它分类。
本期有以下知识点值得关注:
CNS类
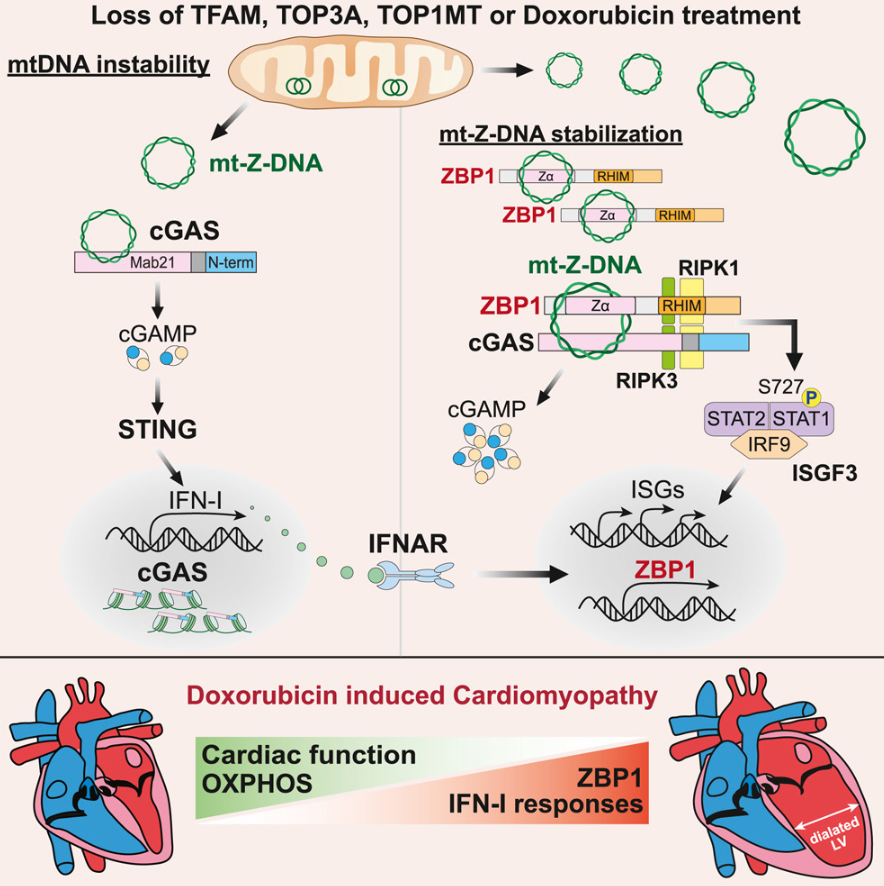
ZBP1 和 cGAS 协同感知线粒体 DNA 促进多柔比星相关心脏毒性
Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell
- 线粒体 DNA (mtDNA) 是先天免疫系统的有效激动剂;然而,mtDNA 的确切免疫刺激特征和胞质核酸传感器的检测动力学仍不清楚。
- 在这里,我们发现线粒体基因组的不稳定性促进了 Z 型 DNA 的积累。 Z-DNA 结合蛋白 1 (ZBP1) 可稳定 Z 型 mtDNA,并使含有 cGAS、RIPK1 和 RIPK3 的胞质复合物成核,以维持 STAT1 磷酸化和 I 型干扰素 (IFN-I) 信号传导。多柔比星是一种一线化疗药物,可在癌症患者中引起频繁的心脏毒性。接触多柔比星后,心肌细胞中的 Z 型 mtDNA、ZBP1 表达和 IFN-I 信号传导升高。引人注目的是,缺乏 ZBP1 或 IFN-I 信号传导的小鼠可以免受阿霉素诱导的心脏毒性。
- 我们的研究结果表明,ZBP1 作为 cGAS 的合作伙伴,维持 IFN-I 对线粒体基因组不稳定性的反应,并强调 ZBP1 作为心力衰竭和其他 mtDNA 应激导致干扰素相关病理的疾病的潜在靶标。
- Bensz/ChatGPT:在特定的条件下,DNA的碱基排列方式可以发生改变,形成了Z型DNA。Z型DNA的特点是碱基之间的排列方式呈现出左旋的形式,形成了一个倒置的双螺旋结构。Z型DNA的形成与多种因素有关,包括DNA序列中的特定序列元素和化学修饰、DNA链的拉伸和张力等。其中,一种称为CpG二核苷酸的特定序列元素在Z型DNA的形成中起到了重要的作用。
肌动蛋白丝的自由端和加帽端的结构
Structures of the free and capped ends of the actin filament. Science
- 肌动蛋白丝 (F-肌动蛋白) 的带刺和尖端是生长和收缩的部位,也是阻止亚基交换的加帽蛋白的目标,包括带刺端的 CapZ 和尖端的原调节蛋白。
- 我们描述了 F-肌动蛋白自由端和加帽端的冷冻电子显微镜结构。自由刺端的末端亚基采用“扁平”F-肌动蛋白构象。 CapZ 的结合对带倒刺的末端进行了微小的改变,但对其自身进行了重大的改变。相比之下,自由尖端的亚基采用“扭曲”单体肌动蛋白(G-肌动蛋白)构象。原调节蛋白结合迫使第二个亚基形成 F-肌动蛋白构象。
- 这些结构揭示了 F-肌动蛋白末端与中间的差异,以及这些差异如何控制亚基添加、解离、加帽以及与末端结合蛋白的相互作用。
油菜素类固醇通过细胞壁和组织力学协调植物细胞层相互作用
Brassinosteroid coordinates cell layer interactions in plants via cell wall and tissue mechanics. Science
- 细胞层之间的生长协调对于大多数多细胞生物的发育至关重要。协调可能是由细胞之间的分子信号和/或机械连接介导的,但基因如何改变各层之间的机械相互作用尚不清楚。
- 在这里,我们表明驱动油菜素类固醇合成的基因至少部分地通过减少机械表皮约束来促进内部组织的生长。我们在水生植物银杏狸中发现了一种油菜素类固醇缺乏的矮化突变体,其内部组织扭曲,可能是由生长缓慢的表皮的机械约束造成的。我们通过证明拟南芥中的油菜素类固醇突变体增强了表皮裂纹的形成(表明组织应力增加)来验证了这一假设。
- 我们提出,通过重塑细胞壁,油菜素类固醇减少表皮约束,展示基因如何通过力学控制各层之间的生长协调。
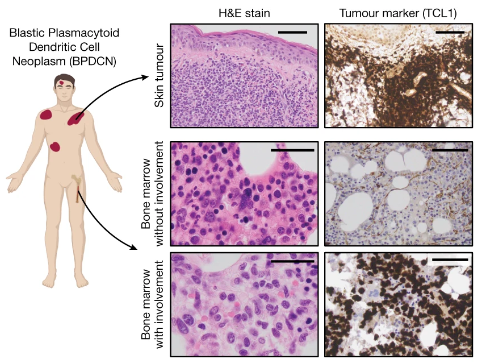
紫外线辐射塑造皮肤树突状细胞白血病转化
Ultraviolet radiation shapes dendritic cell leukaemia transformation in the skin. Nature. full html
一种皮肤内免疫细胞突变导致的特殊肿瘤。挺特别的研究。
- 肿瘤最常由单个解剖生态位内的前体克隆的进展引起。在骨髓中,克隆祖细胞可以恶性转化为急性白血病,或分化为导致外周组织疾病病理的免疫细胞。在骨髓之外,这些克隆可能会暴露于各种组织特异性突变过程,尽管其后果尚不清楚。
- 在这里,我们研究了母细胞性浆细胞样树突状细胞肿瘤(BPDCN)的发展,这是一种不寻常的急性白血病形式,通常表现为皮肤上分离出的恶性细胞。使用肿瘤系统基因组学和单细胞转录组学进行基因分型,我们发现 BPDCN 起源于骨髓中的克隆(癌前)造血前体细胞。
- 我们观察到 BPDCN 皮肤肿瘤首先在暴露于阳光的解剖部位发生,并以紫外线 (UV) 辐射诱导的克隆扩展突变为特征。肿瘤系统发育的重建表明,紫外线损伤可能发生在与恶性转化相关的改变之前,这表明在 BPDCN 发病过程中浆细胞样树突状细胞或定型前体细胞暴露在阳光下。
- 从功能上讲,我们发现 Tet2 功能缺失突变(BPDCN 中最常见的癌前改变)赋予浆细胞样细胞(而非传统的树突状细胞)对紫外线诱导的细胞死亡的抵抗力,这表明 Tet2 具有环境依赖性肿瘤抑制作用。
- 这些发现表明,远处解剖部位的组织特异性环境暴露如何影响癌前克隆向播散性癌症的进化。
Perforin-2 是交叉提呈树突状细胞的内吞逃逸成孔效应子
Perforin-2 is a pore-forming effector of endocytic escape in cross-presenting dendritic cells. Science
英国剑桥 MRC 分子生物学实验室
了解一下该研究的遗传筛选方法
- 在抗病毒和抗肿瘤 T 细胞介导的免疫反应启动期间,树突状细胞 (DC) 在主要组织相容性复合体 (MHC) I 类分子上交叉呈递外源抗原。交叉呈递依赖于树突状细胞中内吞区室的异常“泄漏”,内化的蛋白质由此逃逸到胞质溶胶中,用于蛋白酶体介导的 MHC I 结合肽的生成。鉴于 1 型传统 DC 擅长交叉呈递,我们寻找细胞类型特异性的内吞逃逸效应器。
- 我们设计了一种适合遗传筛选的测定方法,并发现孔形成蛋白穿孔蛋白-2 (Mpeg1)可作为交叉呈递细胞独有的专用效应子。 Perforin-2 被招募到含有抗原的区室中,在那里它经历成熟,释放其成孔结构域。
- Mpeg1−/− 小鼠未能有效地将 CD8+ T 细胞引导至细胞相关抗原,这揭示了穿孔素 2 在交叉呈递过程中抗原进入胞质中的重要作用。
NAC 控制真核生物的新合成蛋白 N 端甲硫氨酸切除
NAC controls cotranslational N-terminal methionine excision in eukaryotes. Science
德国康斯坦茨大学生物学系、瑞士苏黎世联邦理工学院分子生物学和生物物理学研究所
- 由甲硫氨酸氨肽酶 (methionine aminopeptidases, METAP) 共翻译催化的新合成蛋白质的 N 端甲硫氨酸切除是一个重要且普遍保守的过程,在细胞稳态和蛋白质生物合成中发挥着关键作用。然而,METAP 如何与核糖体相互作用以及如何确保其切割特异性尚不清楚。
- 我们发现,在真核生物中,新生多肽相关复合物 (nascent polypeptide–associated complex, NAC) 控制着 METAP1 的核糖体结合。 NAC 使用长而灵活的尾巴招募 METAP1,并为在核糖体隧道出口处形成活性蛋氨酸切除复合物提供平台。这种相互作用模式确保了蛋氨酸从胞质蛋白中有效切除,而靶向内质网的蛋白质则幸免于难。
- 我们的结果提出了一种更广泛的机制来控制蛋白质生物发生因子进入翻译核糖体。
体内神经元尖峰和阈下活动的宽场荧光寿命成像
Wide-field fluorescence lifetime imaging of neuron spiking and subthreshold activity in vivo. Science
美国斯坦福大学物理系
挺神奇的神经活体成像技术
- 电压敏感荧光探针的发展表明荧光寿命作为生物系统中电活动的有前景的读数。现有方法无法达到神经科学应用中电压成像所需的速度和灵敏度。
- 我们证明,宽视场电光荧光寿命成像显微镜(electro-optic fluorescence lifetime imaging microscopy, EO-FLIM)允许以千赫兹帧采集速率进行寿命成像,空间解析活体成年果蝇的动作电位传播和阈下神经活动。
- 单细胞电压记录在 1 kHz 下实现了 <5 皮秒的寿命分辨率。寿命读出受到光子散粒噪声的限制,并且该方法可以强烈抑制运动伪影和技术噪声源。记录揭示了局部跨膜去极化、两种具有不同荧光寿命的尖峰,以及尖峰对外部机械刺激的锁相。
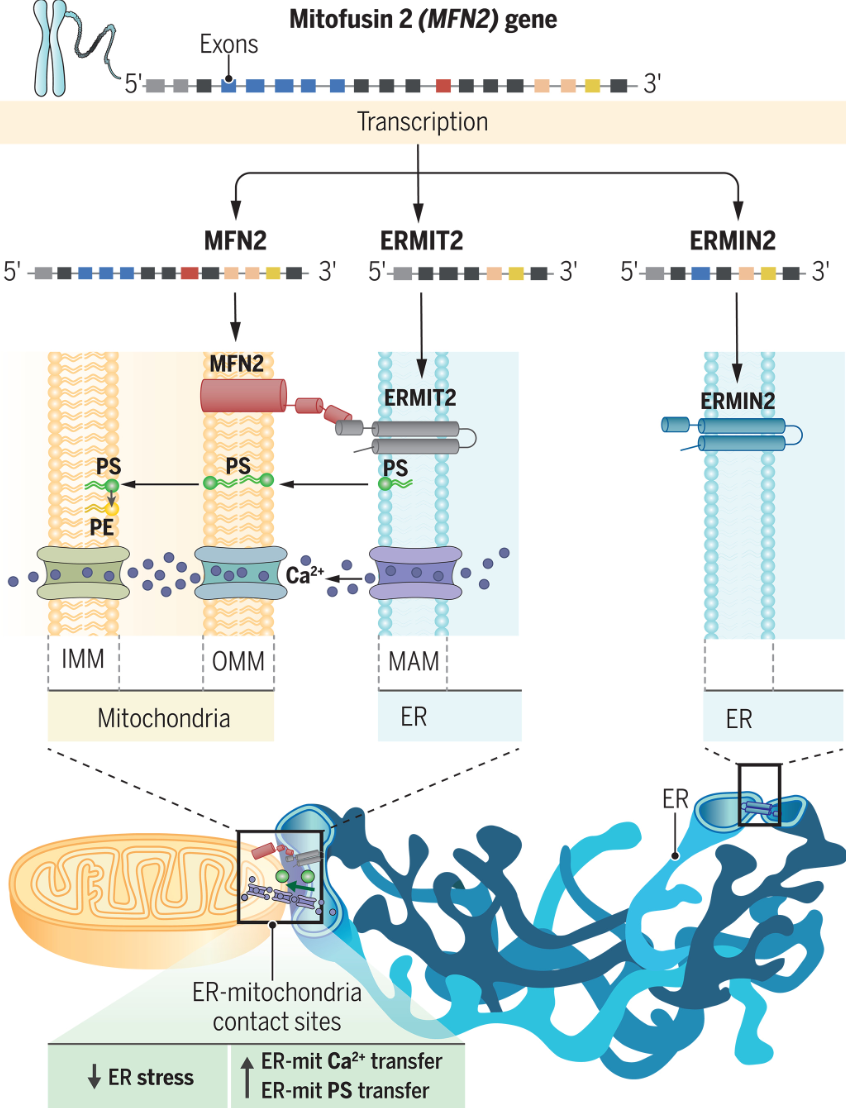
线粒体融合蛋白 2 的剪接变体塑造内质网并将其束缚于线粒体
Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science
挺特点的一个发现
- 在真核细胞的胞浆中,细胞器在膜接触位点(membrane contact sites)紧密并列。膜接触位点为相互作用的细胞器之间共享的代谢和信号级联提供了热点。称为系链的蛋白质桥稳定膜接触位点,以产生代谢物和第二信使交换所必需的微环境。内质网 (ER) 和线粒体在膜接触位点之间的相互作用允许 ER 向线粒体 Ca2+ 和磷脂酰丝氨酸 (PS) 转移,并作为自噬和线粒体裂变起始的位点。在哺乳动物细胞中,线粒体融合蛋白 mitofusin 2 (MFN2) 也起到ER-线粒体结构系绳的作用,但其在内质网膜上的伴侣尚不清楚。
- 单独线粒体上的线粒体融合蛋白之间的相互作用驱动细胞器融合。 MFN2 还可以支持异型细胞器相互作用,如人工 ER 限制性 MFN2 突变体与线粒体上的线粒体融合素物理相互作用的能力所表明的那样。由于 MFN2 具有同型相互作用的倾向,并且线粒体动力学基因选择性剪接成具有不同亚细胞定位和功能的变体,我们假设 ER 上 MFN2 的伴侣可能是 MFN2 基因的剪接变体。我们研究了在人类骨骼肌和小鼠成纤维细胞中发现的称为 ERMIN2 (ER mitofusin 2) 和 ERMIT2 (ER mitofusin 2 tether) 的 MFN2 剪接变体。
- ERMIN2 和 ERMIT2 在多个人体组织中表达,并在驱动 ER 线粒体接近的压力下诱导。我们结合亚细胞定位的成像和生化测定,发现 MFN2 变异仅位于内质网。 ERMIT2 和 MoV-MFN2 富集于 ER-线粒体界面,其 N 和 C 末端暴露于细胞质。这两种变体都无法纠正 Mfn2−/− 线粒体的破坏形态。然而,ERMIN2 对于形成 ER 至关重要,而 ERMIT2 和 MoV-MFN2 对于将其束缚在线粒体上至关重要。位于内质网的 ERMIT2 的卷曲线圈 (CC) 结构域与线粒体线粒体融合蛋白的 CC 区域相互作用,形成一种复合物,该复合物在能量上足够稳定,至少在分子建模模拟中足以束缚这两个细胞器。从功能上讲,ERMIT2 介导的细胞器束缚允许线粒体摄取细胞内 ER 释放的钙。此外,ERMIT2 的细胞器束缚允许在遗传和饮食诱导的 ER-线粒体并置减少模型的肝脏中线粒体摄取 ER 衍生的磷脂。这种束缚最终有助于减轻肝脏内质网应激和炎症。
- 在这项工作中,我们鉴定了单个 MFN2 基因的选择性剪接、ER 限制性变体。这些变体充当 MFN2 在内质网形态和线粒体束缚中线粒体外功能的分子介导者。我们还确定了 MFN2 的束缚功能在肝脏病理生理学中的作用。特别是,在非酒精性脂肪性肝炎的情况下,ERMIT2 可以纠正从 ER 到线粒体的磷脂转移的改变以及随之而来的肝脏窘迫。因此,选择性剪接可以将线粒体塑造机制的功能扩展到线粒体之外,并且改变的内质网线粒体通讯在代谢性肝病的病理学中发挥着关键作用。
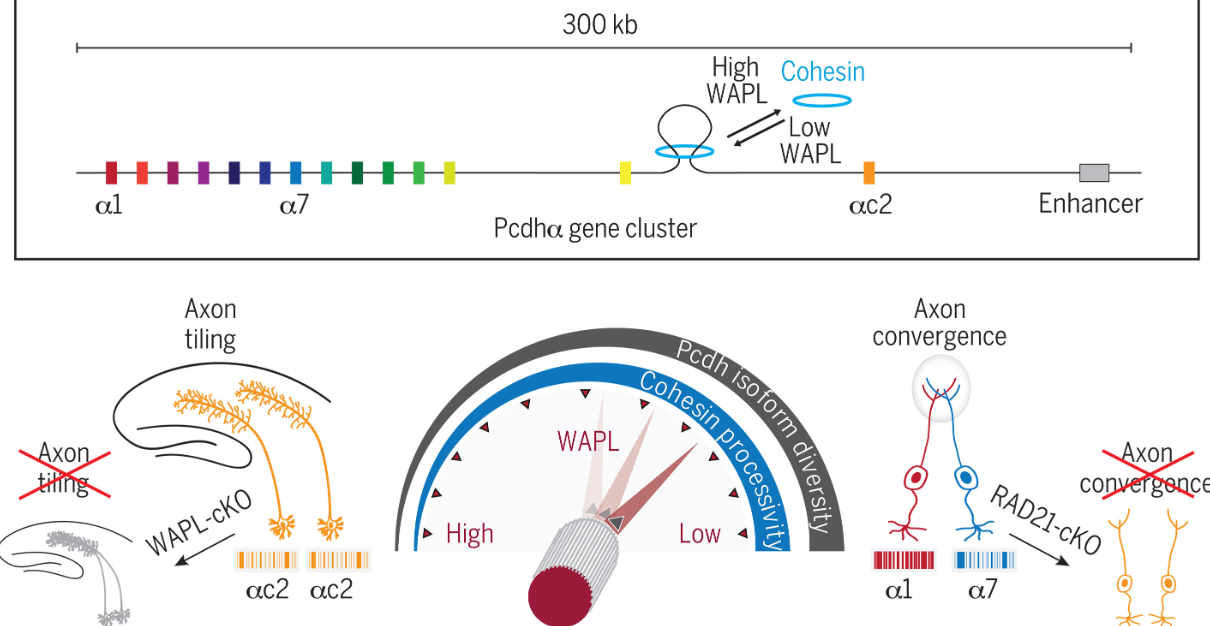
WAPL调节粘连蛋白基因易位从而控制神经元Pcdh基因簇表达的随机性
WAPL functions as a rheostat of Protocadherin isoform diversity that controls neural wiring. Science
美国加州大学-威尔神经科学研究所、旧金山分校神经病学系
我之前对哺乳动物神经元身份识别的背景并不太了解,挺有趣的。这是一篇在写作上十分经典的论文,值得开始写论文的同学好好学习。
- 神经连接模式建立需要单个神经元具有区分自我和非自我的能力。在哺乳动物中,簇状原钙粘蛋白 (Protocadherin, Pcdh) 基因编码细胞表面分子“标识符”(即条形码),允许神经“自我/非自我”区分:来自携带相同 Pcdh 条形码的同一细胞的神经突相互识别并排斥,而携带不同 Pcdh 条形码的细胞则不然。在小鼠中,有 116 个 Pcdh 基因,两条同源染色体上各有 58 个,组织成三个串联排列的簇(α、β 和 γ)。不同的神经类型表达不同的 Pcdh 基因库来指导其接线过程。这种行为最值得注意的例子是血清素能神经元 (5-HT) 中单个 Pcdh 基因的确定性表达和嗅觉感觉神经元 (OSN) 中一些 Pcdh 基因的随机表达。 Pcdhαc2 的确定性表达为 5-HT 提供了单一、共享的条形码。利用这种机制,来自同一细胞的神经突不仅可以识别和排斥自身,还可以识别和排斥来自其他 5-HT 的神经突,从而有利于其轴突在整个大脑中的非重叠平铺投射。相比之下,不同 Pcdh 基因库的随机和组合表达为每个 OSN 提供了独特的条形码。这种条形码多样性的产生使得 OSN 轴突能够汇聚成紧密堆积的重叠结构。这些观察结果提出了一个基本问题:神经元如何选择正确数量(和类型)的 Pcdh 基因来支持其接线需求?
- Pcdh 基因的基因组结构提出了基本的调控挑战:顺式调控机制必须克服 Pcdh 启动子相对于远端增强子的线性排列所带来的基因组邻近偏差。这些机制必须在不同的细胞类型中受到不同的调节。例如,在 5-HT 中,Pcdhαc2 基因的选择存在基因组距离偏差,因为其启动子是 Pcdhα 簇中最靠近增强子的启动子。然而,在 OSN 中,启动子选择中的基因组距离偏差被消除,有利于随机选择。先前的研究表明 DNA 易位酶粘连蛋白通过介导增强子-启动子相互作用来调节 Pcdh 表达。基于这些研究,我们假设粘连蛋白活性的差异调节可能是神经元实现支持其布线模式的 Pcdh 表达程序的潜在机制。
- 通过基因靶向 5-HT 和 OSN 中粘连蛋白复合物的成分,我们揭示了神经类型特异性 Pcdh 表达和轴突行为取决于粘连蛋白及其卸载器 WAPL(wings apart-like protein homolog, 翼分离蛋白同源物)的活性。鉴于 Pcdh 基因的线性排列,5-HT 中的高 WAPL 限制了粘连蛋白易位,从而有利于所有细胞中增强子近端 Pcdhαc2 同工型的表达,并最终将轴突排列限制为平铺模式。 5-HT 中 WAPL 的条件性删除导致 Pcdhαc2 表达丧失和轴突平铺破坏。相比之下,我们发现 OSN 中的低 WAPL 能够使粘连蛋白沿着基因座易位。利用这种机制,粘连蛋白增加了增强子和更远端的 Pcdh 启动子之间接触的可能性,从而能够随机表达更大的 Pcdh 同工型,并最终驱动轴突收敛和嗅觉回路的组装。
- 在 OSN 中,粘连蛋白复合物的重要亚基 Rad21 的条件性消融导致 Pcdh 亚型多样性丧失,并获得增强子近端 Pcdhαc2 基因的偏向表达。因此,Rad21 消融将 OSN 的 Pcdh 表达谱转变为一种让人想起 5HT 的表达谱。 Rad21 缺失导致 Pcdh 多样性丧失,导致 OSN 轴突收敛中断。因此,通过对抗增强子-启动子基因组邻近偏差,WAPL 对粘连蛋白活性的神经类型特异性调节可调节 Pcdh 亚型多样性,并实现不同的神经元布线和电路组装模式。
- 我们提出了一个模型,其中 WAPL 充当 DNA 上粘连蛋白加工性的变阻器,以实现在大脑发育过程中建立不同神经连接模式所需的 Pcdh 基因簇的结构和转录模块化。更广泛地说,我们的数据提出了一种新的基因簇调控原则,其中细胞使用变阻器操作逻辑来克服基因选择中的距离偏差。我们推测变阻器是解决这些基因簇复杂结构带来的挑战的解决方案,这些基因簇产生对细胞和功能多样化至关重要的转录多样性。
基于哈扎狩猎采集者的研究揭示工业化对人类肠道微生物组的影响
Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Nature
样本特别。
- 肠道微生物组调节免疫和代谢健康。人类微生物组数据偏向工业化人群,限制了我们对非工业化微生物组的理解。
- 在这里,我们对来自坦桑尼亚哈扎狩猎采集者以及尼泊尔和加利福尼亚的比较人群的 351 个粪便样本进行了超深度宏基因组测序。
- 我们恢复了 91,662 个细菌、古生菌、噬菌体和真核生物的基因组,其中 44% 不存在于现有的统一数据集中。我们发现了 124 个在工业化人群中消失的肠道驻留物种,并强调了哈扎肠道微生物组与原位复制率、选择特征和菌株共享相关的独特方面。
- 人们发现工业化肠道微生物富含与氧化应激相关的基因,这可能是微生物组适应炎症过程的结果。
- 这种对哈扎人肠道微生物组的无与伦比的看法提供了宝贵的资源,扩大了我们对能够在人类肠道定殖的微生物的理解,并澄清了工业化生活方式引起的广泛扰动。
癌细胞丢失Y 染色体后可诱导适应性免疫抑制
Y chromosome loss in cancer drives growth by evasion of adaptive immunity. Nature
挺有趣的研究,思路也不复杂。该现象也并不适用于女性肿瘤。该研究也并未完全解释肿瘤细胞Y染色体丢失是如何影响体内的抗肿瘤免疫的。
了解一下CRISPR-Cas9靶向删除整条Y染色体的方法。
- 在多种癌症类型中观察到 Y 染色体 (LOY) 丢失,包括 10-40% 的膀胱癌,但其临床和生物学意义尚不清楚。
- 在这里,我们使用基因组学和转录组学研究报告,LOY 与膀胱癌患者的不良预后相关。我们对自然发生的 LOY 突变膀胱癌细胞以及通过 CRISPR-Cas9 靶向删除 Y 染色体的细胞进行了深入研究。
- Y阳性(Y+)和Y阴性(Y–)肿瘤在体外生长相似,而Y−肿瘤以T细胞依赖性方式在免疫活性宿主中比Y+肿瘤更具侵袭性。高维流式细胞术分析表明,Y− 肿瘤会促进肿瘤微环境中 CD8+ T 细胞的显着功能障碍或耗竭。使用单核 RNA 测序和人类膀胱癌的空间蛋白质组学评估验证了这些发现。值得注意的是,与 Y+ 肿瘤相比,Y− 肿瘤在小鼠和癌症患者中对抗 PD-1 免疫检查点阻断疗法的反应增强。
- 总之,这些结果表明,具有 LOY 突变的癌细胞会改变 T 细胞功能,促进 T 细胞耗竭,并使它们对 PD-1 靶向免疫疗法敏感。这项工作提供了对 LOY 突变的基本生物学和改善癌症免疫疗法的潜在生物标志物的见解。
GFP DNA 模拟物的复杂 3D 结构
- 大量研究表明 RNA 分子如何采用复杂的三维 (3D) 结构。相比之下,DNA 是否可以自组装成复杂的 3D 折叠,能够独立于蛋白质或 RNA 伴侣进行复杂的生物化学,仍然是个谜。
- Lettuce 是一种体外进化的 DNA 分子,可结合并激活 4 个源自 GFP 的条件荧光团。为了将先前的荧光 RNA、GFP 和其他荧光蛋白的结构研究扩展到 DNA,我们通过 X 射线晶体学和低温电子显微镜对生菜-荧光团复合物进行了表征。
- 结果表明,53 个核苷酸的 DNA 采用四路连接 (4WJ) 折叠。与 4WJ RNA 中常见的典型 L 形或 H 形结构不同,Lettuce 的四个茎形成两个同轴堆叠,它们共线堆积以形成荧光团结合的中央 G-四链体。这种折叠通过堆叠、广泛的核碱基氢键结合——包括通过不寻常的对角线堆叠的碱基连接 DNA 的主要同轴堆叠的连续层——以及单价和二价阳离子的协调来稳定。总体而言,该结构比许多同等大小的 RNA 更紧凑。
- Lettuce 展示了 DNA 如何在不使用类似 RNA 的三级相互作用的情况下形成精细的三维结构,并表明核酸组织的新原理将从复杂的 DNA 分析中得出。
衰老黑素细胞发出的信号使毛发生长过度活跃
Signalling by senescent melanocytes hyperactivates hair growth. Nature. full html
- Niche 信号维持干细胞长时间静止或短暂激活它们以进行适当的再生。改变平衡的生态位信号可导致再生障碍。人类的黑色素细胞皮肤痣经常表现出过度的毛发生长,表明毛发干细胞过度活跃。
- 在这里,使用 nevi 的遗传小鼠模型,我们表明衰老黑色素细胞的真皮簇驱动上皮毛发干细胞退出静止状态并改变它们的转录组和组成,从而有效地促进毛发更新。痣黑色素细胞激活一个独特的分泌蛋白组,富含信号因子。
- 骨桥蛋白是主要的痣信号因子,对于诱导毛发生长是必要且充分的。注射骨桥蛋白或其基因过表达足以诱导小鼠毛发生长,而骨桥蛋白或 CD44(其上皮毛细胞上的同源受体)的种系和条件性缺失可挽救由真皮痣黑素细胞诱导的毛发生长增强。
- 骨桥蛋白在人类毛状痣中过度表达,可刺激人类毛囊的新生长。尽管衰老细胞的广泛积累,例如衰老或基因毒性应激,对组织的再生能力是有害的,但我们表明,衰老细胞簇发出的信号可以有效增强相邻完整干细胞的活性并刺激组织更新。
- 这一发现将衰老细胞及其分泌蛋白组确定为再生障碍中有吸引力的治疗靶点。
PD-1 维持 CD8 T 细胞对皮肤新抗原的耐受性
PD-1 maintains CD8 T cell tolerance towards cutaneous neoantigens. Nature
美国耶鲁大学医学院免疫生物学系
这个动物模型挺特别的
- 健康个体的外周 T 细胞库包含自身反应性 T 细胞。 PD-1 等检查点受体被认为能够通过自身反应性 CD8 T 细胞的缺失或无反应来诱导外周耐受。然而,该模型受到接受检查点抑制剂治疗的癌症患者中免疫相关不良事件发生频率较高的挑战。
- 在这里,我们开发了一种小鼠模型,其中表皮中 T 细胞抗原的皮肤特异性表达引起具有效应基因表达谱的抗原特异性 CD8 T 细胞的局部浸润。在这种情况下,PD-1 通过阻止组织浸润的抗原特异性效应 CD8 T 细胞 (1) 获得功能齐全的致病性分化状态,(2) 分泌大量效应分子,以及 ( 3) 接触表皮抗原表达细胞。
- 在没有 PD-1 的情况下,表皮抗原表达细胞被抗原特异性 CD8 T 细胞消除,导致局部病理学。对两名患有皮肤苔藓样免疫相关不良事件的患者的皮肤活检进行的转录组分析显示,病变和非病变皮肤中均存在克隆扩增的效应 CD8 T 细胞。
- 因此,我们的数据支持外周 T 细胞耐受模型,其中 PD-1 允许抗原特异性效应 CD8 T 细胞与组织中抗原表达细胞共存,而无需免疫病理学。
调节抗肿瘤免疫的 B 细胞特异性检查点分子
B-cell-specific checkpoint molecules that regulate anti-tumour immunity. Nature
美国哈佛医学院和布莱根妇女医院恒大免疫疾病中心
近年来B细胞调控抗肿瘤免疫的机制研究越来越多了。研究思路还是相对简单的。
- B 细胞在抗肿瘤免疫中的作用仍存在争议,因此,免疫疗法的重点是靶向 T 细胞和自然杀伤细胞以抑制肿瘤生长。
- 在这里,我们利用高通量流式细胞术以及在 B16F10 黑色素瘤生长过程中对 B 细胞进行批量和单细胞 RNA 测序以及 B 细胞受体测序分析,鉴定出了在荷瘤小鼠淋巴结引流中随时间的变化的特异性扩增的 B 细胞子集。
- 不断扩大的 B 细胞亚群表达细胞表面分子 T 细胞免疫球蛋白和粘蛋白结构域 1(TIM-1,由 Havcr1 编码)和独特的转录特征,包括多种共抑制分子,如 PD-1、TIM-3、TIGIT 和LAG-3。尽管有条件地删除 B 细胞上的这些共抑制分子对肿瘤负荷影响很小或没有影响,但选择性删除 B 细胞中的 Havcr1 既能显着抑制肿瘤生长,又能增强效应 T 细胞反应。 TIM-1 的缺失增强了 B 细胞中的 1 型干扰素反应,从而增强了 B 细胞的活化并增加了抗原呈递和共刺激,从而导致肿瘤特异性效应 T 细胞的扩增增加。
- 我们的结果表明,对表达 TIM-1 的 B 细胞进行操作可以激活适应性免疫的第二臂,从而促进抗肿瘤免疫并抑制肿瘤生长。
宽松的靶向规则有助于 PIWI 蛋白沉默转座子
Relaxed targeting rules help PIWI proteins silence transposons. Nature. full html
挺特别的发现。我对PIWI相互作用RNA的了解不多,有空可看看全文。
- 在真核生物中,小 RNA 向导(例如小干扰 RNA 和 microRNA)指导 AGO 进化枝 Argonaute 蛋白调节基因表达并保护基因组免受外部威胁。只有动物才能产生 Argonaute 蛋白的第二个分支:PIWI 蛋白。 PIWI 蛋白使用 PIWI 相互作用 RNA (piRNA) 来抑制互补转座子转录本。理论上,转座子可以通过降低 piRNA 互补性的靶位点突变来逃避沉默。
- 在这里,我们报告说,与 AGO 蛋白不同,PIWI 蛋白可以有效地切割仅与其 piRNA 向导部分配对的转录本。对小鼠和海绵 PIWI 蛋白的靶标结合和切割的检查表明,PIWI 切片可以容忍与任何靶标核苷酸的错配,包括那些位于可裂解磷酸盐侧翼的核苷酸。即使是规范的种子配对对于 PIWI 结合或裂解来说也是可有可无的,这与植物和动物 AGO 不同,植物和动物 AGO 需要从种子到断裂键后的核苷酸不间断的靶标配对。因此,PIWI 蛋白比 AGO 蛋白更能靶向新获得的或快速分化的内源转座子,而无需依赖新的小 RNA 指导。相反,PIWI 切片的最低要求足以避免宿主 RNA 的无意沉默。
- 我们的结果证明了 PIWI 相对于 AGO 蛋白在保护基因组免受转座子侵害方面的生物学优势,并解释了为什么 piRNA 途径在动物进化中得以保留。
损伤相关EGFR信号可防止嵌合皮肤中的 Ras 突变细胞扩张
Injury prevents Ras mutant cell expansion in mosaic skin. Nature. full html
这个发现听上去有点奇怪。如果该现象成立,HrasG12V/+ 细胞怎么在周围有健康上皮细胞的情况下发展为肿瘤呢?
- 健康皮肤是野生型和突变型克隆的嵌合体。虽然损伤可以与突变的 Ras 家族蛋白合作促进肿瘤发生,但基因嵌合皮肤的后果尚不清楚。
- 在这里,我们发现,受伤后野生型细胞会抑制致癌 Ras 诱导的异常生长。 HrasG12V/+ 和 KrasG12D/+ 细胞在未受伤的镶嵌组织中胜过野生型细胞,但由于增殖的野生型细胞比例增加,它们在受伤后的扩张受到阻止。
- 从机制上讲,我们表明,与 HrasG12V/+ 细胞不同,野生型细胞对 EGFR 配体的自分泌和旁分泌分泌有反应,EGFR 通路的这种差异激活解释了损伤修复过程中的竞争性转换。通过药物或遗传方法抑制 EGFR 信号传导会减少损伤后分裂的野生型细胞的比例,从而导致 HrasG12V/+ 细胞的扩增。通过细胞周期抑制剂 p21 的组成性缺失增加野生型细胞的增殖,即使在没有损伤的情况下也能抵消 HrasG12V/+ 细胞的扩增。
- 因此,损伤在改变基因嵌合皮肤中致癌细胞和野生型细胞之间的竞争平衡方面发挥着作用。
组蛋白去甲基化酶 KDM5D 上调导致结肠癌的性别差异
Histone demethylase KDM5D upregulation drives sex differences in colon cancer. Nature
挺特别的发现
- 性别对癌症的发病率、谱系和结果具有深远的影响,但性别差异的分子和遗传基础尚不明确,推测归因于 X 染色体基因和性激素。这种性别差异在结直肠癌(CRC)中尤为突出,男性的转移率和死亡率更高。
- 采用编码致癌突变体 KRASG12D 的诱导转基因以及 Apc 和 Trp53 肿瘤抑制因子(称为 iKAP)的条件无效等位基因设计的小鼠 CRC 模型显示,特别是在患有致癌突变体 KRAS (KRAS*) CRC 的男性中,转移率更高,结果更差。
- 综合跨物种分子和转录组分析确定 Y 染色体基因组蛋白去甲基化酶 KDM5D 是由 KRAS* 介导的 STAT4 转录因子激活驱动的转录上调基因。 KDM5D 依赖性染色质标记和转录组变化显示上皮细胞紧密连接调节因子和MHC-I成分受到抑制。 iKAP 癌细胞中 Kdm5d 的缺失增加了紧密连接的完整性,降低了细胞侵袭性并增强了 CD8+ T 细胞对癌细胞的杀伤力。相反,用 Kdm5d 转基因工程化以在 iAP 癌细胞中提供组成型 Kdm5d 表达的 iAP 小鼠显示出体内更具侵袭性肿瘤的倾向增加。
- 因此,KRAS*-STAT4 介导的 Y 染色体 KDM5D 上调通过破坏癌细胞粘附特性和肿瘤免疫,极大地促进了 KRAS* CRC 的性别差异,为患有癌症的男性降低转移风险提供了可行的治疗策略。
小鼠早期发育中核糖体占用的单细胞定量
Single-cell quantification of ribosome occupancy in early mouse development. Nature. full html
美国德克萨斯大学奥斯汀分校分子生物科学系
- 翻译调控对于早期哺乳动物胚胎发育至关重要。然而,以前的研究仅限于批量测量,排除了翻译调节的精确确定,包括等位基因特异性分析。
- 在这里,为了应对这一挑战,我们开发了一种新的微流体等速电泳 (ITP) 方法——RIBOsome profiling via ITP (Ribo-ITP),并表征了小鼠早期发育过程中单个卵母细胞和胚胎的翻译。我们将差异翻译效率确定为调节参与中心体组织和 RNA 的 N6-甲基腺苷修饰的基因的关键机制。
- 据我们所知,我们的高覆盖率测量首次对早期发育中的等位基因特异性核糖体参与进行了分析。这些导致发现了合子 RNA 与核糖体的阶段特异性差异结合,并降低了表现出等位基因偏向表达的转录本的翻译效率。通过将我们的测量与蛋白质组学数据相结合,我们发现生发泡期卵母细胞中的核糖体占据是受精卵中蛋白质丰度的主要决定因素。
- 总之,Ribo-ITP 方法将通过从包括单细胞在内的超低输入样本提供高覆盖率和高分辨率核糖体占据测量来实现众多应用。
T 细胞受体α 多样性的起源和进化延展性
Origin and evolutionary malleability of T cell receptor α diversity. Nature. full html
看不太懂。 正文较简短,有时间可以看看。
- 脊椎动物适应性免疫系统的淋巴细胞获得了从种系中的分裂基因组装数十亿个功能性抗原受体的能力。这些受体表现出特异性;与先天系统广泛调节的受体不同,例如,B 细胞表达的抗体 (Ig) 可以准确地区分有机酸的两种对映体,而 T 细胞受体 (TCR) 可以可靠地识别其肽抗原中的单个氨基酸替代物。在发育中的淋巴细胞中,抗原受体基因由一组相对较小的种系编码遗传元件组装而成,这一过程称为 V(D)J 重组。由准随机体细胞多样化引起的一些抗原受体的潜在自身反应性被几种强大的控制机制抑制。几十年来,科学家们一直对体细胞多样化抗原受体的进化起源感到困惑。目前尚不清楚,在这一机制诞生之初,免疫学上有益的扩大的受体多样性是如何与新出现的破坏性自我识别的风险进行交换的。
- 在这里,我们探讨了这样的假设:在早期脊椎动物中,标记重组元件末端的序列微同源性成为选择的关键目标,决定了 RAG 介导的重组过程中产生的 DNA 双链断裂的基于非同源末端连接的修复的结果。
- 我们发现,在有颌脊椎动物的主要进化枝中,TCRα 库多样性最好是通过此类序列微同源性的物种特异性程度来解释。因此,重排元件的种系序列组成的选择成为决定体细胞产生的抗原受体多样性程度的主要因素。
后脑模块差异化地改变单个丘脑神经元的活动以协调运动
- 看似简单的行为,例如拍蚊子或看路标,都涉及多个身体部位的精确协调。协调运动的神经控制被广泛认为需要将所需的整体位移转换为每个身体部位的位移。
- 在这里,我们揭示了小鼠凝视系统中实现的不同逻辑。刺激上丘 (SC) 会引起具有定型位移的头部运动,但会引起具有定型端点的眼球运动。
- 这是通过单个 SC 神经元实现的,其分支轴突支配髓质和脑桥中的模块,分别驱动具有刻板位移的头部运动和具有刻板端点的眼球运动。因此,单个神经元指定不同身体部位的端点和位移的混合,而不是整体位移,不同身体部位的位移在不同的解剖阶段计算。
- 我们的研究建立了一种解开运动层次结构的方法,并确定了协调运动和最终姿势的逻辑。
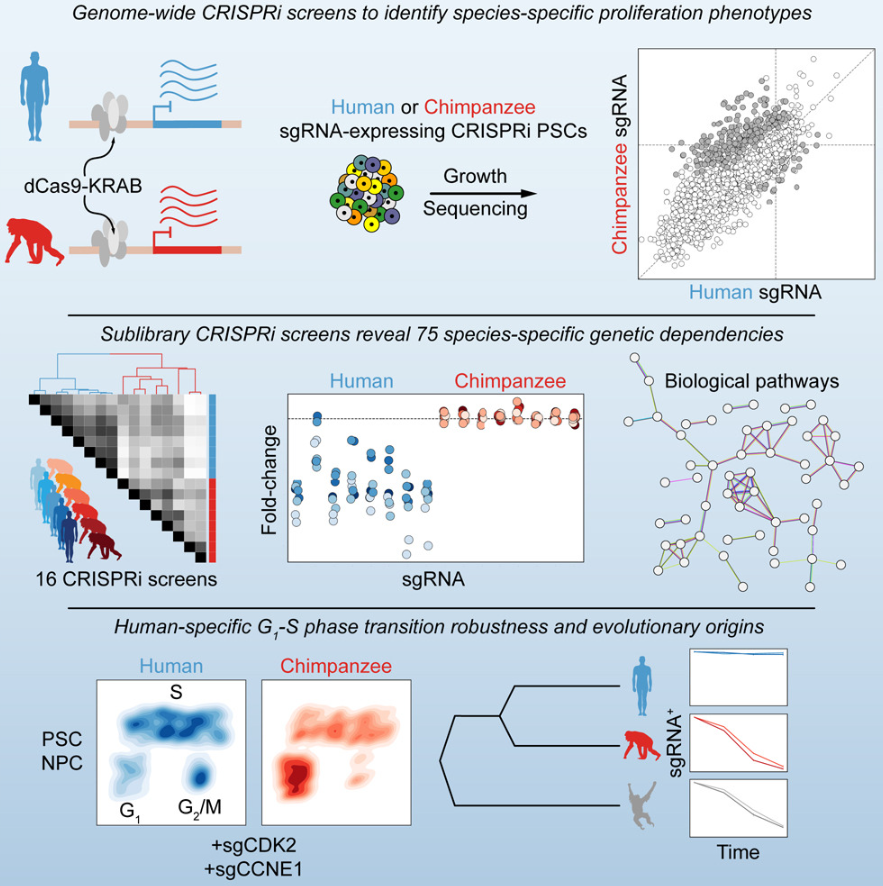
比较人类和黑猩猩干细胞的遗传依赖性
Comparative landscape of genetic dependencies in human and chimpanzee stem cells . Cell. full html
- 对类人猿的比较研究为了解我们的进化历史提供了一个窗口,但在古人类进化过程中出现的细胞差异的程度和身份在很大程度上仍未得到探索。我们建立了一种比较功能丧失方法来评估人类细胞是否表现出独特的遗传依赖性。
- 通过在人类和黑猩猩多能干细胞中进行全基因组 CRISPR 干扰筛选,我们鉴定了 75 个对细胞增殖具有物种特异性影响的基因。这些基因包含连贯的过程,包括细胞周期进程和溶酶体信号传导,通过与猩猩细胞进行比较,我们确定这些基因是人类衍生的。
- 人类特有的对 CDK2 和 CCNE1 耗竭的稳健性在神经祖细胞和大脑类器官中持续存在,支持 G1 期长度假说作为人类大脑扩张的潜在进化机制。
- 我们的研究结果表明,人类细胞的进化变化重塑了必需基因的格局,并建立了一个系统地揭示物种之间潜在的细胞和分子差异的平台。
肿瘤免疫类
肿瘤相关纤维化损害非小细胞肺癌的免疫监视和对免疫检查点阻断的反应
Tumor-associated fibrosis impairs immune surveillance and response to immune checkpoint blockade in non-small cell lung cancer. Sci Transl Med
- 非小细胞肺癌(NSCLC)是癌症相关死亡的主要原因。免疫检查点阻断提高了许多非小细胞肺癌患者的生存率,但大多数患者未能获得长期获益。了解导致 NSCLC 免疫监视减少的因素对于改善患者预后至关重要。
- 在这里,我们发现人类 NSCLC 存在大量纤维化,这与 T 细胞浸润减少相关。在小鼠 NSCLC 模型中,纤维化的诱导导致肺癌进展加快、T 细胞免疫监视受损以及免疫检查点阻断功效失效。与这些变化相关,我们观察到纤维化导致树突状细胞数量和功能受损以及巨噬细胞表型改变,这可能导致免疫抑制。
- 在癌症相关成纤维细胞中,表达 Col13a1 的群体内的明显变化表明这些细胞产生趋化因子来招募巨噬细胞和调节性 T 细胞,同时限制树突状细胞和 T 细胞的招募。通过转化生长因子-β受体信号传导来靶向纤维化,克服了纤维化的影响,增强了 T 细胞反应,并提高了免疫检查点阻断的功效,但仅限于化疗的情况下。
- 总之,这些数据表明 NSCLC 中的纤维化导致免疫监视减少和对检查点封锁的反应较差,并强调抗纤维化治疗是克服免疫治疗耐药性的候选策略。
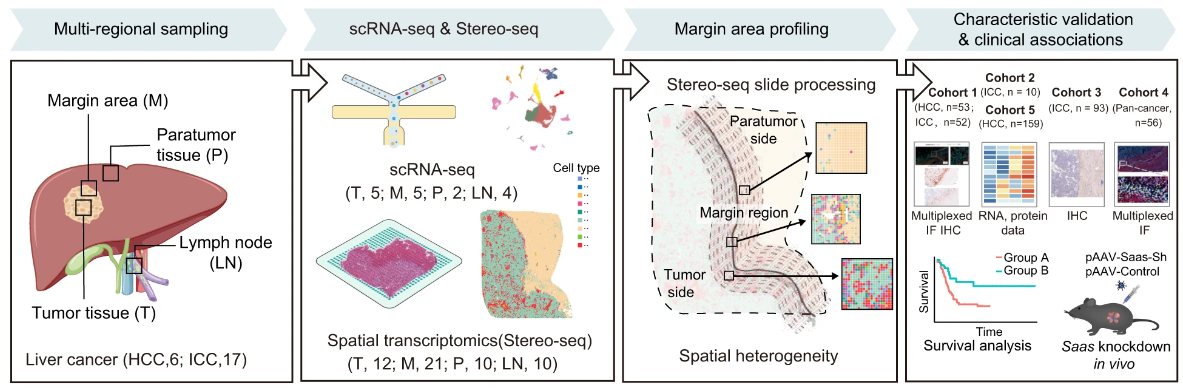
Stereo-seq 识别人肝癌中的“侵袭区”
An invasive zone in human liver cancer identified by Stereo-seq promotes hepatocyte-tumor cell crosstalk, local immunosuppression and tumor progression. Cell Res. full html
复旦大学中山医院中山华大精准医疗中心
- 解剖和了解癌症生态系统,特别是肿瘤边缘周围的生态系统,对肿瘤细胞浸润和侵袭具有重要意义,对于探索肿瘤转移机制和开发有效的新疗法至关重要。
- 使用纳米级分辨率的新型肿瘤边界扫描和数字化模型 – SpaTial 增强型 REsolution 组学测序 (Stereo-seq),我们在肝癌患者的肿瘤边界周围确定了一个 500 µm 宽的区域,称为“侵入区”。我们在该区域检测到强烈的免疫抑制、代谢重编程和严重受损的肝细胞。我们还发现了一个受损肝细胞亚群,其血清淀粉样蛋白 A1 和 A2(统称为 SAA)表达增加,位于肿瘤旁边界附近。
- 邻近恶性细胞中CXCL6的过度表达可以诱导附近肝细胞中JAK-STAT3通路的激活,从而导致这些肝细胞中SAA的过度表达。此外,侵袭区肝细胞过度表达和分泌SAA可能导致巨噬细胞的募集和M2极化,进一步促进局部免疫抑制,可能导致肿瘤进展。
- 另外五个独立的原发性和继发性肝癌患者队列 (n = 423) 的临床关联分析表明,侵袭区 SAA 过度表达的患者预后较差。使用小鼠肝肿瘤模型进行的进一步体内实验证实,肝细胞中编码 SAA 的基因的敲除减少了肿瘤边界周围巨噬细胞的积累,并延迟了肿瘤的生长。
- 人类癌症患者中新型侵袭区的识别和表征不仅增加了对肿瘤侵袭和转移机制的重要理解,而且还可能为开发晚期肝癌和其他实体瘤的新型治疗策略铺平道路。
MHC-II相关免疫耐受促进乳腺癌淋巴结转移
有点出忽意料
- 肿瘤引流淋巴结 (TDLN) 对于肿瘤抗原特异性 T 细胞的生成和有效的抗癌免疫反应非常重要。然而,TDLN 通常是转移的主要部位,导致免疫抑制和更糟糕的结果。
- 通过跨物种单细胞 RNA 测序分析,我们确定了乳腺癌进展和淋巴结转移 (LNM) 过程中癌细胞异质性、可塑性和免疫逃避的特征。在小鼠和人类中,淋巴结中的一部分癌细胞表现出 MHC II 类 (MHC-II) 基因表达升高。 MHC-II+ 癌细胞缺乏共刺激分子表达,导致 TDLN 中调节性 T 细胞 (Treg) 扩增和 CD4+ 效应 T 细胞减少。 MHC-II 的基因敲除减少了 LNM 和 Treg 的扩张,而 MHC-II 反式激活因子 Ciita 的过度表达则使 LNM 恶化并导致 Treg 过度扩张。
- 这些发现表明癌细胞 MHC-II 表达促进 TDLN 中的转移和免疫逃避。
肿瘤和局部淋巴组织的相互作用决定高级别浆液性卵巢癌的预后
Tumor and local lymphoid tissue interaction determines prognosis in high-grade serous ovarian cancer. Cell Rep Med
中规中矩的研究框架
- 三级淋巴结构(Tertiary lymphoid structure, TLS)与拷贝数驱动的肿瘤(包括高级别浆液性卵巢癌(HGSOC))的预后相关,尽管 TLS 的功能及其与 HGSOC 拷贝数改变的相互作用尚未完全了解。
- 在当前的研究中,我们证实 TLS 高 HGSOC 患者表现出明显更好的无进展生存期 (PFS)。我们发现 HGSOC 肿瘤中 TLS 的存在与 B 细胞成熟和细胞毒性肿瘤特异性 T 细胞激活和增殖有关。此外,IL15和CXCL10的拷贝数丢失可能会限制HGSOC中TLS的形成;还提出了可能失调 TLS 功能的基因列表。
- 最后,开发了基于放射组学的特征来预测 TLS 的存在,该特征可以独立预测 HGSOC 患者和免疫检查点抑制剂 (ICI) 治疗的非小细胞肺癌 (NSCLC) 患者的 PFS。
- 总的来说,我们揭示了 TLS 协调肿瘤内 B 细胞和 T 细胞对 HGSOC 肿瘤的反应,而癌症基因组的进化则抵消了 TLS 的形成和功能。
临床类
免疫治疗时代的临终系统肿瘤治疗:种族、保险和实践环境的作用
End-of-Life Systemic Oncologic Treatment in the Immunotherapy Era: The Role of Race, Insurance, and Practice Setting. J Clin Oncol
- 目的:已证明在生命末期 (EOL) 接受抗肿瘤全身治疗会损害患者和护理人员的体验,增加住院次数、重症监护病房和急诊科的使用以及驾车费用;然而,这些比率并没有下降。为了了解抗肿瘤 EOL 全身治疗的影响因素,我们探讨了其与实践和患者层面因素的关联。
- 方法:我们纳入了来自真实世界电子健康记录的去识别化数据库中的患者,这些患者自 2011 年开始接受诊断的晚期或转移性癌症的全身治疗,并在 2015 年至 2019 年之间的 4 年内死亡。我们评估了 EOL (死亡前14天) 全身治疗在 30 岁至 2019 年的使用情况。我们将治疗分为三个子类别:单独化疗、化疗和免疫治疗联合治疗以及免疫治疗(有/无靶向治疗),并使用多变量混合水平逻辑回归估计患者和实践因素的条件比值比 (OR) 和 95% CI 。
- 结果:在来自 150 个诊所的 57,791 名患者中,19,837 名患者在死亡前 30 天内接受了全身治疗。我们观察到 36.6% 的白人患者、32.7% 的黑人患者、43.3% 的商业保险患者和 37.0% 的医疗补助患者接受了 EOL 全身治疗。白人患者和拥有商业保险的患者比黑人患者或拥有医疗补助的患者更有可能接受 EOL 全身治疗。与学术中心的治疗相比,社区诊所的治疗与接受 30 天系统 EOL 治疗的几率更高(调整后的 OR,1.51)。我们观察到不同实践中 EOL 系统治疗率存在很大差异。
- 结论:在大量现实世界人群中,EOL 系统治疗率与患者种族、保险类型和实践环境相关。未来的工作应该研究导致这种使用模式的因素及其对下游护理的影响。
BLM 过表达作为 PARP 抑制剂耐药 BRCA 突变卵巢癌 CHK1 抑制剂反应的预测生物标志物
BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor-resistant BRCA-mutant ovarian cancer. Sci Transl Med. full html
- 聚(ADP-核糖)聚合酶抑制剂(PARPis)改变了乳腺癌基因(BRCA)突变的高级别浆液性卵巢癌(HGSC)的治疗模式。然而,大多数患者最终会对 PARPis 产生耐药性,这凸显了对改进治疗策略的需求尚未得到满足。
- 通过高通量药物筛选,我们鉴定出共济失调性毛细血管扩张症和 rad3 相关蛋白/检查点激酶 1 (CHK1) 通路抑制剂具有细胞毒性,并进一步在HGSC 细胞和异种移植小鼠模型验证了 CHK1 抑制剂 (CHK1i) prexasertib 在 PARPi 敏感和耐药的 BRCA 突变体中的活性。 CHK1i 单一疗法可诱导 DNA 损伤、细胞凋亡和肿瘤尺寸减小。
- 我们对 BRCA 突变 HGSC 患者进行了 prexasertib 的 2 期研究 (NCT02203513)。该治疗耐受性良好,但在既往接受过 PARPi 治疗的患者中,客观缓解率为 6%(17 例中有 1 例;部分缓解)。探索性生物标志物分析表明,复制应激和叉稳定性与 CHK1i 的临床益处相关。特别是,在从 CHK1i 获得持久益处的患者中观察到 Bloom 综合征 RecQ 解旋酶 (BLM) 和细胞周期蛋白 E1 (CCNE1) 过度表达或拷贝数增加/扩增。先前接受过 PARPi 治疗的 BRCA 突变患者中的 BRCA 回复突变与 CHK1i 耐药性无关。
- 我们的研究结果表明,复制叉相关基因应作为 BRCA 突变 HGSC 患者 CHK1i 敏感性的生物标志物进行进一步评估。
整合治疗中改良格拉斯哥预后评分和影像学来预测转移性肾细胞癌的反应和结果
- 在免疫肿瘤学时代,仅靠影像学似乎不足以捕捉治疗反应,因为接受免疫疗法治疗的疾病稳定的患者具有广泛的临床结果。对于能够结合成像评估治疗反应和结果的补充(理想情况下具有成本效益)标记物的需求尚未得到满足。
- 探讨基于 C 反应蛋白和白蛋白的改良Glasgow预后评分 (mGPS) 的纵向变化是否可以预测转移性肾细胞癌 (mRCC) 患者的反应和结果。设计、设置和参与者:这项事后分析于 2022 年 10 月至 2023 年 4 月进行,在 2 项随机临床试验中评估了治疗中 mGPS 对接受阿特珠单抗(加贝伐单抗)或舒尼替尼治疗的 mRCC 患者的预后和预测性能:3 期 IMmotion151 研究(发现队列)和 2 期 IMmotion150 研究(验证队列)。结局是研究者根据实体瘤疗效评估标准 (RECIST) 1.1 版评估的无进展生存期 (PFS) 和生存分析的总生存期 (OS)。为了比较治疗中 mGPS 与放射学分期的预后价值,我们使用了独立审查委员会 (IRC-RECIST) 评估的 RECIST,以确保高数据质量。
- 在 IMmotion151 发现队列中的 915 名 mRCC 患者中,861 名患者可使用基线 mGPS,691 名患者可使用治疗中 mGPS。IMmotion150 验证队列包括 305 名 mRCC 患者,199 名患者可评估治疗中 mGPS。在 IMmotion150 研究中,治疗中 mGPS 最早可在治疗开始后 6 周预测结果,从而为早期治疗调整打开了窗口。
- 在这两项临床试验中,无论首次分期时影像评估的治疗反应如何,治疗中的 mGPS 都提供了有价值的预后信息。值得注意的是,在疾病控制亚组中,与 IRC-RECIST(适用于 611 名患者;治疗期间 mGPS 的 C 指数为 0.651 [95% CI,0.588-0.714] IRC-RECIST 为 0.574 [95% CI,0.528-0.619]。
- 这些数据支持除了放射学分期之外还整合治疗中 mGPS 进行更全面和以患者为中心的治疗监测的概念,以低成本改善 mRCC 患者的临床护理。
用于在资源有限的环境中检测 HPV16 和 HPV18 DNA 的集成等温核酸扩增测试
An integrated isothermal nucleic acid amplification test to detect HPV16 and HPV18 DNA in resource-limited settings. Sci Tran Med
低成本临床转化研究,值得关注。
- 高危人乳头瘤病毒 (HPV) DNA 检测被广泛认为是最敏感的宫颈癌筛查方法,但在资源有限、宫颈癌负担最高的地区,其可用性有限。最近,HPV DNA 检测已被开发用于资源有限的环境,但它们对于广泛使用而言仍然过于昂贵,并且需要的仪器通常仅限于集中实验室。
- 为了帮助满足全球对低成本宫颈癌筛查的需求,我们开发了一种针对 HPV16 和 HPV18 DNA 的原型、从样本到答案(sample-to-answer)的即时护理(point-of-care)测试。我们的测试依赖于等温 DNA 扩增和侧流检测,这两种技术可以减少对复杂仪器的需求。在美国资源丰富的环境和资源匮乏的莫桑比克中,我们分图将所有测试组件集成到一个低成本、可制造的平台里,并自行收集临床样本来评估集成测试的性能。
- 我们证明了每次测试 1000 个 HPV16 或 HPV18 DNA 拷贝的临床相关检测限。该测试需要六个用户步骤,在 45 分钟内产生结果,并且可以由经过最低限度培训的人员使用台式仪器和微型离心机进行。预计每次测试成本 < 5 美元,预计仪器成本 < 1000 美元。
- 这些结果表明了从样本到答案、即时 HPV DNA 检测的可行性。由于纳入了其他 HPV 类型,该检测有可能填补分散且全球可及的宫颈癌筛查的关键空白。
其它类
NKX6-2驱动IPMN的胃性分化
- 胰腺导管内乳头状粘液性肿瘤(IPMN)是胰腺导管腺癌(PDAC)的真正前驱病变。 IPMN 最常见的亚型具有胃小凹型上皮,这些低度粘液性肿瘤是具有高度不典型增生和癌症的 IPMN 的先兆。 IPMN 胃分化的分子基础尚不清楚,尽管识别这种惰性表型的驱动因素可能有机会阻止向高级 IPMN 和癌症的进展。
- 我们对一组 IPMN 进行了空间转录组学,然后进行了正交和跨物种验证研究,确定转录因子 NKX6-2 作为低级 IPMN 中胃细胞身份的关键决定因素。 NKX6-2 表达的丧失是 IPMN 进展的一致特征,而小鼠 IPMN 系中 NKX6-2 的重新表达概括了上述胃转录程序和腺体形态。
- 我们的研究确定 NKX6-2 是一种以前未知的转录因子,在 IPMN 发病机制中驱动惰性胃分化。
局部EGFR-抗体阻断小分子用药减少EGFRi的皮肤毒性
Preventing skin toxicities induced by EGFR inhibitors by topically blocking drug-receptor interactions. Sci Transl Med
- 表皮生长因子受体(EGFR)抑制剂用于治疗许多晚期上皮癌,但在大多数接受治疗的患者中会引起严重的皮肤毒性。这些副作用导致患者生活质量恶化并影响抗癌治疗。目前针对这些皮肤毒性的治疗策略侧重于减轻症状,而不是预防引起毒性的最初触发因素。
- 在这项研究中,我们开发了一种化合物和方法,通过在毒性部位阻断药物而不减少到达肿瘤的全身剂量来治疗“靶向”皮肤毒性。我们首先筛选了能够有效阻断抗EGFR单克隆抗体与EGFR结合的小分子,并确定了一个潜在的候选药物SDT-011。计算机对接预测 SDT-011 与 EGFR 上的相同残基相互作用,这些残基对于 EGFR 抑制剂西妥昔单抗和帕尼单抗的结合很重要。
- SDT-011 与 EGFR 的结合降低了西妥昔单抗与 EGFR 的结合亲和力,并且可以重新激活角质形成细胞系、离体西妥昔单抗处理的整个人类皮肤和注射 A431 的小鼠中的 EGFR 信号传导。局部应用特定的小分子,并通过源自可生物降解纳米颗粒的缓释系统递送,该系统可穿透毛囊和皮脂腺,其中 EGFR 高度表达。
- 我们的方法有可能减少 EGFR 抑制剂引起的皮肤毒性。
粘附层粘连蛋白-1 和 IV 型胶原蛋白诱导 Ca2+ 微结构域的形成并敏化小鼠 T 细胞
Adhesion to laminin-1 and collagen IV induces the formation of Ca2+ microdomains that sensitize mouse T cells for activation. Science Signaling
- 在免疫反应期间,T 细胞穿过内皮并穿过细胞外基质 (ECM),从血管壁迁移到发炎组织。整合素促进 T 细胞与内皮细胞和 ECM 蛋白结合。
- 在这里,我们报告在没有 T 细胞受体 (TCR)/CD3 刺激的情况下观察到的 Ca2+ 微结构域是由 ECM 蛋白粘附触发的初始信号事件,增加了原代小鼠 T 细胞对激活的敏感性。对 ECM 蛋白 IV 型胶原和层粘连蛋白 1 的粘附以依赖于激酶 FAK、磷脂酶 C (PLC) 和所有三种肌醇 1,4,5-三磷酸受体 (IP3R) 亚型的方式增加了 Ca2+ 微结构域的数量,并促进了转录因子 NFAT-1 的核转位。
- 数学模型预测,粘附依赖性 Ca2+ 微区的形成需要 2 至 6 个 IP3R 和 ORAI1 通道的协同活动,以实现 ER-质膜连接处 Ca2+ 浓度的增加,这是实验观察到的,并且需要 SOCE。此外,通过整体 Ca2+ 反应和 NFAT-1 核易位评估,粘附依赖性 Ca2+ 微区对于 TCR 诱导的 IV 型胶原上 T 细胞活化的程度非常重要。
- 因此,与 IV 型胶原蛋白和层粘连蛋白-1 的粘附通过涉及 Ca2+ 微结构域形成的机制使 T 细胞敏化,并且阻断这种低水平的敏化可减少 TCR 接合时的 T 细胞活化。
机器学习/组学类
基于人结直肠癌类器官的药物再利用筛选及机制分析
Drug repurposing screening and mechanism analysis based on human colorectal cancer organoids. Protein Cell
比较常规的套路
- 结直肠癌(CRC)是一种高度异质性的癌症,探索新的治疗方案是一个需要解决的紧迫问题。在这里,我们建立了人类结直肠癌肿瘤衍生的类器官,它很好地代表了原始肿瘤的形态和分子异质性。
- 为了有效地识别结直肠癌的重新利用药物,我们开发了一个强大的基于类器官的药物筛选系统。通过结合重新利用的药物库和基于计算的药物预测,测试了 335 种药物,并鉴定了 34 种具有抗 CRC 作用的药物。
- 更重要的是,我们对药物反应进行了详细的转录组分析,并将药物反应特征分为五种代表性模式:分化诱导、生长抑制、代谢抑制、免疫反应促进和细胞周期抑制。候选药物的抗癌活性在已建立的基于患者的类器官异种移植(PDOX)体内系统中得到进一步验证。我们发现fedratinib、trametinib 和bortezomib 表现出有效的抗癌作用。此外,还评估了体外类器官和体内成对 PDOX 之间药物反应特征的一致性和不一致。
- 我们的研究为药物发现提供了一种创新方法,药物反应的代表性转录组特征为开发结直肠癌的新型临床治疗方法提供了宝贵的资源。
胃癌多组学分析
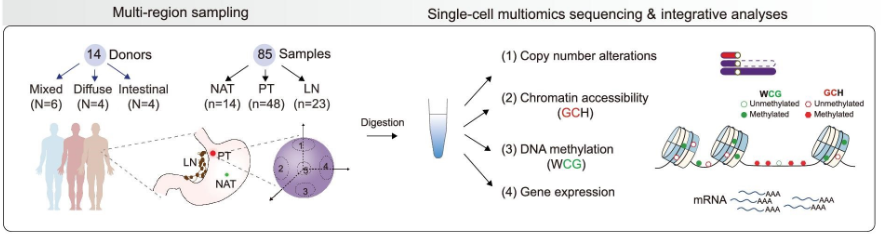
Integrative single-cell multiomics analyses dissect molecular signatures of intratumoral heterogeneities and differentiation states of human gastric cancer. Natl Sci Rev. full html
- 人类胃癌是一种高度致命的疾病,但潜在的多组学分子特征仍不清楚。在这里,我们对人类胃癌进行了多区域采样、并行单细胞多组学测序和综合分析。
- 我们发现了胃癌细胞常见的转录组改变,例如与正常胃功能相关的基因的异常下调和KRT7、PI3、S100A4等的上调。令人惊讶的是,正常胃功能中高表达的基因异常且普遍上调。结直肠上皮细胞也在癌细胞中被发现,这可能部分受到启动子染色质可及性和 DNA 甲基化水平的调节。
- 我们揭示了胃癌的单细胞 DNA 甲基化组图谱,并鉴定了候选 DNA 甲基化生物标志物,例如 TMEM240 和 HAGLROS 的高甲基化启动子,以及 TRPM2-AS 和 HRH1 的低甲基化启动子。此外,在单细胞水平上系统地揭示了遗传谱系、DNA甲基化和转录组簇之间的关系。我们发现DNA甲基化异质性主要存在于癌细胞的不同遗传谱系之间。此外,我们发现,在同一患者的原发肿瘤中,分化状态较差的癌细胞的DNA甲基化水平往往高于分化状态较好的癌细胞,尽管仍低于正常胃上皮细胞。分化状态较差的癌细胞也普遍下调 MUC1 表达和免疫相关通路,并且 CD8+ T 细胞浸润较差。
- 我们的研究利用综合单细胞多组学分析剖析了人胃癌瘤内异质性和分化状态的分子特征。
基于生物库级别数据的遗传图谱快速估计
Fast inference of genetic recombination rates in biobank scale data. Genome Res. full pdf
- 虽然整个基因组的重组事件率(遗传图谱)是遗传研究的基础,但目前的大多数研究仅使用一张标准图谱。有证据表明遗传图谱存在群体差异,因此估计特定群体的图谱很有意义。虽然最近生物样本库规模数据的可用性提供了这样的机会,但当前的方法在利用非常大的样本量方面效率不高。最准确的方法仍然是基于连锁不平衡 (LD) 的方法,该方法仅适用于几百个样品。
- 在这项工作中,我们提出了一种快速且节省内存的方法,用于根据群体基因分型数据估计遗传图谱。我们的方法 FastRecomb 利用高效的位置 Burrows-Wheeler 变换 (PBWT) 数据结构将 IBD 段边界计数为潜在的重组事件。我们使用 PBWT 块来避免成对匹配的冗余计数。此外,我们使用面板平滑技术来减少错误和最近突变带来的噪音。
- 通过仿真,我们发现 FastRecomb 在 10k 分辨率下实现了估计图与地面实况之间的相关系数方面最先进的性能。这主要是因为 FastRecomb 可以有效地利用包含数十万个单倍型的大型面板。与此同时,其他方法缺乏处理此类数据的效率。
- 我们相信 FastRecomb 的进一步完善将为遗传学界提供更准确的遗传图谱。
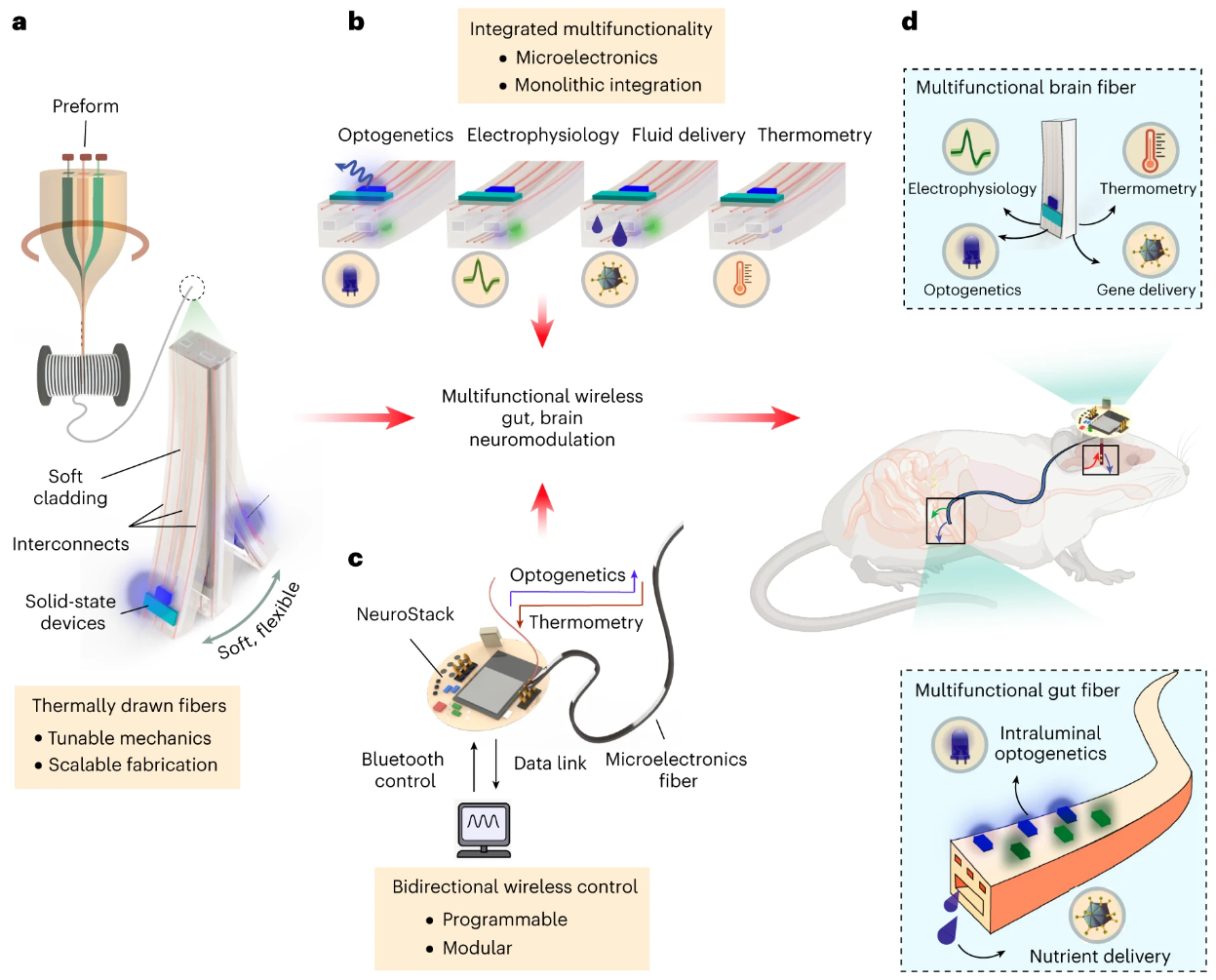
多功能微电子纤维能够无线调节肠道和大脑神经回路
Multifunctional microelectronic fibers enable wireless modulation of gut and brain neural circuits. Nat Biotechnol. full html
挺特别的发明
- 由于缺乏适合在行为过程中探测大脑和周围器官神经生理学的植入设备,阻碍了对大脑-内脏内感受信号传导的理解。
- 在这里,我们描述了多功能神经接口,它将热拉聚合物基纤维的可扩展性和机械多功能性与用于大脑和肠道等多种器官的微电子芯片的复杂性结合起来。
- 我们的方法使用米长的连续纤维,可以将光源、电极、热传感器和微流体通道集成在一个微型材料里。与定制的控制模块配合使用,光纤可以无线传输用于光遗传学的光并传输用于生理记录的数据。我们通过调节小鼠大脑中的中边缘奖励通路来验证这项技术。然后,我们将这些纤维应用在解剖学上具有挑战性的肠腔中,并演示了对指导进食行为的感觉上皮细胞的无线控制。最后,我们证明来自肠腔的迷走神经传入的光遗传学刺激足以在不受束缚的小鼠中引起奖赏表型。
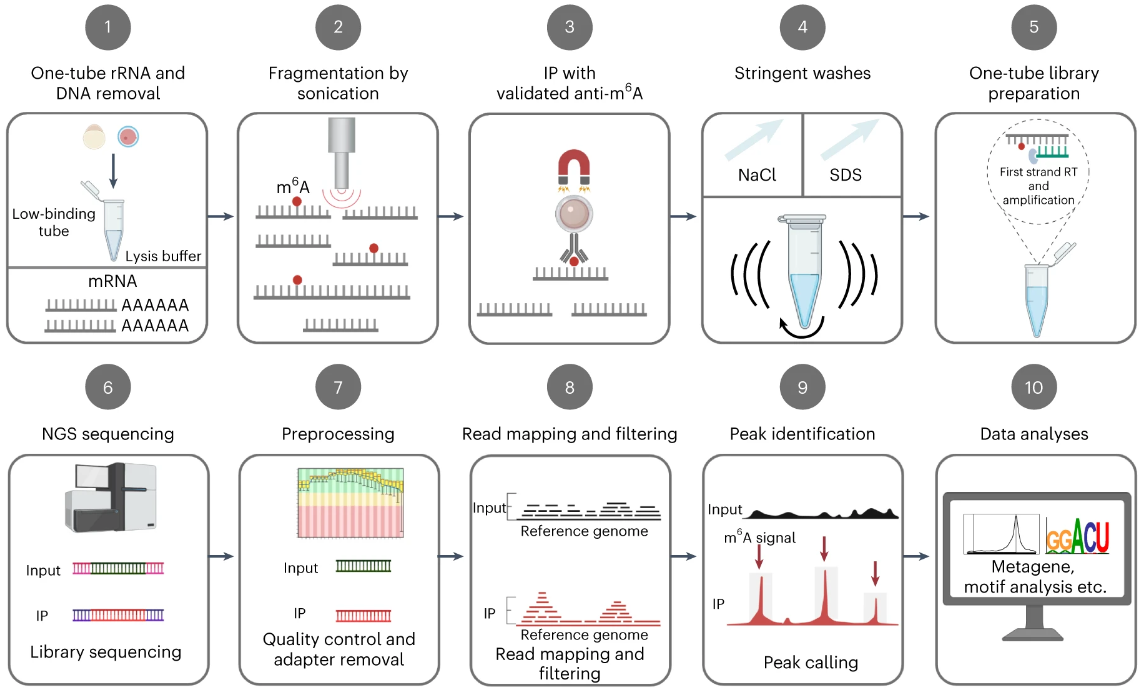
picoMeRIP–seq:单细胞m6A检测
Single-cell m6A mapping in vivo using picoMeRIP-seq. Nat Biotechnol. full html
- 目前的 N6-甲基腺苷 (m6A) 作图方法需要大量 RNA 或仅限于培养细胞。
- 通过优化样品回收率和信噪比,我们开发了皮克级 m6A RNA 免疫沉淀和测序 (picoMeRIP-seq),用于使用标准实验室设备在单细胞和稀有细胞类型中体内研究 m6A。
- 我们对聚 (A) RNA 和胚胎干细胞以及单个斑马鱼受精卵、小鼠卵母细胞和胚胎的滴定进行 m6A 作图基准测试。
气道微生物组与肺功能损伤
广东省疾病预防控制中心、华南师范大学生命科学学院生态科学研究所
样本稀有
- 暴露于环境污染会影响呼吸系统健康。气道微生物生态系统在暴露与呼吸系统健康相互作用中的作用仍不清楚。
- 在这里,通过中国广东省范围内的慢性阻塞性肺疾病监测计划,我们对 1,651 名家庭成员的诱导痰液中的细菌 (n = 1,651) 和真菌 (n = 719) 分类单元和宏基因组 (n = 1,128) 进行了人群调查。
- 我们发现吸烟和较高的 PM2.5 浓度分别通过细菌和真菌群落的介导与肺功能损伤相关,并且暴露与增强的微生物界相互作用相关,类似于慢性阻塞性肺疾病中观察到的模式。奈瑟菌的富集与高呼吸道症状负担的风险增加 2.25 倍相关,同时曲霉菌的升高与职业污染有关。
- 我们开发了一种基于微生物组的个体化健康指数,该指数与暴露、呼吸道症状和疾病相关,具有对全球数据集的潜在普适性。
- 我们的结果可以为环境风险预防提供信息,并指导利用气道微生物组的干预措施。
卵巢早衰致病性变异的外显率
Penetrance of pathogenic genetic variants associated with premature ovarian insufficiency. Nat Med
- 卵巢早衰 (POI) 影响 1% 的女性,是导致不孕的主要原因。它通常被认为是一种单基因疾病,文献中描述了约 100 个基因的致病变异。我们试图利用英国生物银行 104,733 名女性的外显子组序列数据,系统地评估这些基因变异的外显率,其中 2,231 名女性 (1.14%) 报告称自然绝经年龄在 40 岁以下。
- 我们发现有限的证据支持任何先前报道的常染色体显性效应。对于先前报道的 POI 基因的几乎所有杂合效应,我们甚至排除了适度的外显率,在生殖健康女性中发现了 99.9%(13,708 中的 13,699)的所有蛋白质截短变异。我们在几个基因中发现了单倍体不足效应的证据,包括TWNK(绝经提前1.54年,P = 1.59 × 10-6)和SOHLH2(绝经提前3.48年,P = 1.03 × 10-4)。
- 总的来说,我们的结果表明,对于绝大多数女性来说,POI 并不是由先前报道的或目前在临床诊断小组中评估的基因中的常染色体显性变异引起的。我们的发现加上之前的研究表明,大多数 POI 病例本质上可能是寡基因或多基因的,这对未来的临床遗传学研究以及受 POI 影响的家庭的遗传咨询具有重要意义。
扩展空间转录组学
Expansion spatial transcriptomics. Nat Methods. full pdf & supplementary materials
美国斯坦福大学生物工程系
- 基于捕获阵列的空间转录组学方法已广泛用于解析组织中的基因表达;然而,它们的空间分辨率受到阵列密度的限制。
- 在这里,我们提出了扩展空间转录组学,通过在使用增强协议捕获整个聚腺苷酸化转录组之前清除和扩展组织来克服这一限制。这种方法使我们能够实现更高的空间分辨率,同时保持高文库质量。
- 我们使用小鼠大脑样本证明了这一点。
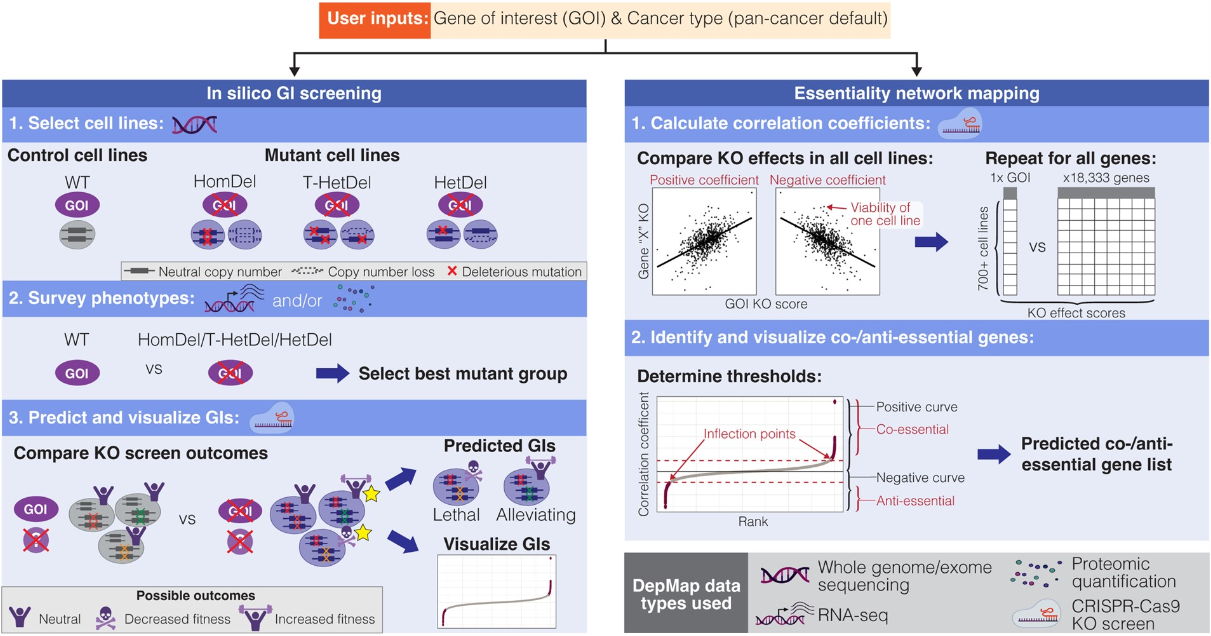
In silico 遗传相互作用和必要性网络分析
GRETTA: an R package for mapping in silico genetic interaction and essentiality networks . Bioinformatics
加拿大不列颠哥伦比亚大学迈克尔·史密斯基因组科学中心
- 绘制人类细胞系中的遗传相互作用和必要性网络已分别用于识别携带特定遗传改变的细胞的脆弱性并将新功能与基因相关联。破译这些网络的体外和体内遗传筛选是资源密集型的,限制了可分析样品的通量。
- 我们提供了一个 R 包,称为 Genetic interRaction and EssenTiality neTwork mApper (GRETTA)。 GRETTA 是一种使用公开数据进行计算机遗传相互作用筛选和重要性网络分析的易用工具,仅需要基本的 R 编程知识。
- Singularity 容器也可从 https://cloud.sylabs.io/library/ytakemon/gretta/gretta 获取。
细胞形状研究框架FlowShape
Cell shape characterization, alignment and comparison using FlowShape. Bioinformatics
- 细胞形状受到严格控制,反映了包括肌动球蛋白活性、粘附特性、细胞分化和极化在内的重要过程。因此,将细胞形状与遗传和其他扰动联系起来具有丰富的信息。然而,目前使用的大多数细胞形状描述符仅捕获简单的几何特征,例如体积和球形度。我们提出了 FlowShape,这是一个以完整且通用的方式研究细胞形状的新框架。
- 在我们的框架中,细胞形状是通过测量形状曲率并将其以共形方式映射到球体上来表示的。接下来,球面上的这个单一函数通过级数展开来近似:球谐函数分解。分解有利于许多分析,包括形状对齐和统计单元形状比较。新工具用于对细胞形状进行完整的通用分析,使用早期秀丽隐杆线虫胚胎作为模型案例。我们区分并表征了七细胞阶段的细胞。
- 我们还设计了一个过滤器来识别细胞形状上的突起,以突出细胞中的片状伪足。此外,该框架还可用于识别 Wnt 通路基因敲除后的任何形状变化。首先使用快速傅里叶变换对细胞进行最佳对齐,然后计算平均形状。接下来量化条件之间的形状差异并与经验分布进行比较。
- 我们通过开源软件包 FlowShape 提出了核心算法的高性能实现,以及表征、对齐和比较细胞形状的例程。重新创建结果所需的数据和代码可在 https://doi.org/10.5281/zenodo.7778752 上免费获取。
宏蛋白质组分析方法MetaNovo
MetaNovo: An open-source pipeline for probabilistic peptide discovery in complex metaproteomic datasets. PLoS Comput Biol
- 微生物组研究为复杂微生物生态系统的代谢相互作用提供了重要的新见解,涉及人类疾病发病机制、农业和气候变化等多种领域。通常观察到 RNA 和蛋白质表达数据集之间的相关性较差,因此很难从宏基因组数据中准确推断微生物蛋白质的合成。此外,基于质谱的宏蛋白质组分析通常依赖于基于蛋白质鉴定先验知识的集中搜索序列数据库,这些数据库可能并不代表一组样品中存在的所有蛋白质。宏基因组 16S rRNA 测序仅针对细菌成分,而全基因组测序充其量只是表达蛋白质组的间接测量。
- 在这里,我们描述了一种新颖的方法,MetaNovo,它结合了现有的开源软件工具来执行可扩展的从头序列标签匹配,并使用一种新颖的算法来对整个 UniProt 知识库进行概率优化,从而为直接在目标诱饵搜索中创建定制的序列数据库。蛋白质组水平,无需事先预期样品组成或宏基因组数据生成即可进行宏蛋白质组分析,并且与标准下游分析流程兼容。
- 我们将 MetaNovo 与 MetaPro-IQ 管道在 8 个人类粘膜-管腔界面样本上发布的结果进行了比较,与使用匹配的宏基因组发现的结果相比,肽和蛋白质鉴定数量相当,许多共享肽序列和类似的细菌分类分布序列数据库,但同时比以前的方法鉴定了更多的非细菌肽。 MetaNovo 还针对匹配的宏基因组和全基因组序列数据库工作流程对已知微生物组成的样本进行了基准测试,为预期的分类群提供了更多的 MS/MS 鉴定,并改进了分类学表征,同时还强调了之前描述的基因组测序质量问题之一生物体,并在没有事先预期的情况下识别实验样品污染物。
- 通过串联质谱数据直接估计微生物组样品的分类和肽水平信息,MetaNovo 能够同时识别元蛋白质组样品中生命所有领域的肽,无需搜索精选的序列数据库。我们表明,MetaNovo 质谱元蛋白质组学方法比当前定制或匹配的基因组序列数据库搜索的金标准方法更准确,可以在没有事先预期的情况下识别样品污染物,并基于复杂质量的潜力,深入了解以前未识别的元蛋白质组学信号光谱测定宏蛋白质组数据不言而喻。
大肠杆菌菌落的全细胞建模能够量化抗生素反应中的单细胞异质性
Whole-cell modeling of E. coli colonies enables quantification of single-cell heterogeneity in antibiotic responses. PLoS Comput Biol
美国斯坦福大学生物工程系
比较小众
- 抗生素耐药性对人类健康构成越来越大的风险,因为目前的抗生素对耐药性日益增强的病原菌正在失去功效。特别值得关注的是多重耐药菌株的出现,这种菌株在革兰氏阴性菌(如大肠杆菌)中迅速出现。大量工作已经证实抗生素耐药机制取决于表型异质性,这可能是由抗生素耐药基因的随机表达介导的。这种分子水平表达与由此产生的群体水平之间的联系是复杂且多尺度的。因此,为了更好地理解抗生素耐药性,需要新的机制模型来反映单细胞表型动态和群体水平异质性,作为一个整体。
- 在这项工作中,我们试图通过建立我们之前在“全细胞”建模方面的经验来架起单细胞和群体规模建模的桥梁,这种方法整合了生物过程的数学和机械描述,以概括实验观察到的整个细胞的行为。
- 为了将全细胞建模扩展到“全菌落”规模,我们将全细胞大肠杆菌模型的多个实例嵌入到动态空间环境的模型中,从而使我们能够在包含以下内容的云上运行大型并行模拟:先前全细胞模型的所有分子细节以及在共享环境中生长的菌落的许多相互作用效应。由此产生的模拟用于探索大肠杆菌对两种具有不同作用机制的抗生素(四环素和氨苄青霉素)的反应,使我们能够识别亚代表达的基因,例如 β-内酰胺酶 ampC,它极大地促进了显着的结果。稳态周质氨苄青霉素的细胞差异,是决定细胞存活的重要因素。
DeepSTF:转录因子结合位点预测
DeepSTF: predicting transcription factor binding sites by interpretable deep neural networks combining sequence and shape. Brief Bioinform
有train.py脚本,可了解下。
- 精确定位转录因子结合位点 (TFBS) 对于理解转录调控过程和研究细胞功能至关重要。尽管已经创建了几种深度学习算法来预测 TFBS,但模型的内在机制和预测结果很难解释。预测性能仍有改进的空间。
- 我们推出 DeepSTF,这是一种独特的深度学习架构,用于通过整合 DNA 序列和形状轮廓来预测 TFBS。我们首次在 TFBS 预测方法中使用改进的 Transformer 编码器结构。 DeepSTF使用堆叠卷积神经网络(CNN)提取DNA高阶序列特征,而通过结合改进的变压器编码器结构和双向长短期记忆(Bi-LSTM)来提取丰富的DNA形状轮廓,最后导出更高阶的DNA形状轮廓。 顺序序列特征和代表性形状轮廓被集成到通道维度中以实现准确的TFBS预测。
- 对 165 个 ENCODE 染色质免疫沉淀测序 (ChIP-seq) 数据集的实验表明,DeepSTF 在预测 TFBS 方面远远优于几种最先进的算法,我们解释了 Transformer 编码器结构的有用性以及使用序列特征和形状的组合策略配置文件捕获多个依赖项并学习基本功能。此外,本文还探讨了 DNA 形状特征预测 TFBS 的重要性。
HiCLift:染色质接触基因组坐标转换
HiCLift: A fast and efficient tool for converting chromatin interaction data between genome assemblies. Bioinformatics
美国伊利诺伊州芝加哥西北大学范伯格医学院生物化学和分子遗传学系
基因组版本转换
- 随着人类参考基因组质量的不断提高以及越来越多个人基因组的产生,基因组组装之间基因组坐标的转换在许多综合和比较研究中至关重要。虽然已经开发了用于线性基因组信号的工具(例如 ChIP-Seq),但不存在将基因组组装转换为染色质相互作用数据的工具,尽管三维(3D)基因组组织在基因调控和疾病中很重要。
- 在这里,我们推出了 HiCLift,这是一种快速高效的工具,可以将 Hi-C 和 Micro-C 等染色质接触的基因组坐标从一个装配转换为另一个装配,包括最新的 T2T 基因组。与直接将原始读数重新映射到不同基因组的策略相比,HiCLift 的运行速度平均快 42 倍(小时与天),同时输出几乎相同的接触矩阵。
- 更重要的是,由于HiCLift不需要重新映射原始读数,它可以直接转换人类患者样本数据,而原始测序读数有时很难获取或无法获得。
- 可用性:HiCLift 已在 https://github.com/XiaoTaoWang/HiCLift 上公开提供。
Nanomonsv基于长读测序检测体细胞SV
Precise characterization of somatic complex structural variations from tumor/control paired long-read sequencing data with nanomonsv. Nucleic Acids Res
日本东京国立癌症中心研究所基因组分析平台开发部
- 我们展示了我们的新型软件 nanomonsv,用于使用肿瘤和具有单碱基分辨率的匹配对照长读长测序数据来检测体细胞结构变异(SV)。
- Nanomonsv 当前版本包括两个检测模块,Canonical SV 模块和 Single Breakend SV 模块。使用来自三种癌症及其匹配的淋巴母细胞系的肿瘤/对照配对长读长测序数据,我们证明 Canonical SV 模块可以识别可通过短读长技术捕获的体细胞 SV,其精度和召回率比现有方法更高。
- 此外,我们还开发了一个工作流程来对移动元素插入进行分类,同时阐明其深入属性,例如 5′ 截断、内部倒位以及 3′ 转导的源位点。此外,单断裂末端 SV 模块能够检测只能通过长读长识别的复杂 SV,例如涉及高度重复着丝粒序列的 SV,以及 LINE1 和病毒介导的重排。
- 总之,我们应用于癌症长读长测序数据的方法可以揭示体细胞 SV 的各种特征,并将有助于更好地理解体细胞 SV 的突变过程和功能后果。
通过强化学习建模和预测癌症克隆进化
Modeling and predicting cancer clonal evolution with reinforcement learning. Genome Res
- 癌症是由进化过程产生的,该过程通常会在同一肿瘤内产生具有不同突变组的多个克隆。准确地模拟这一过程是理解和预测癌症进化的关键。
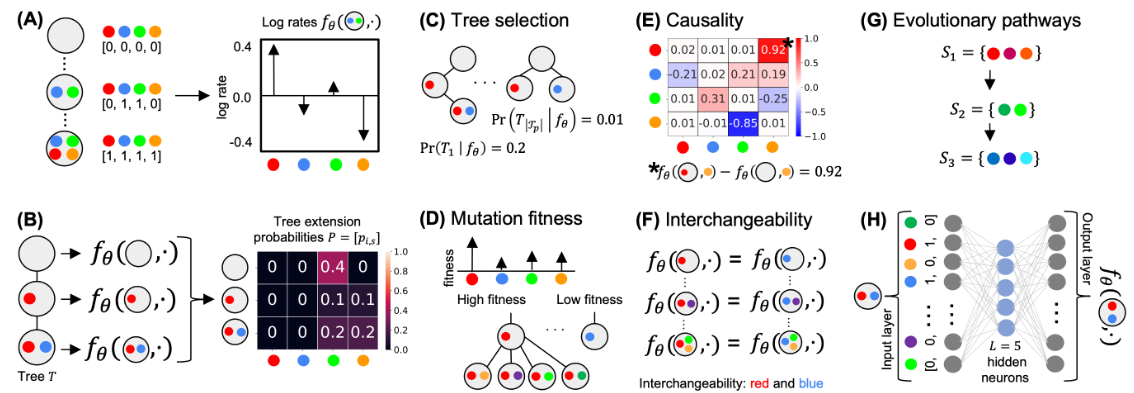
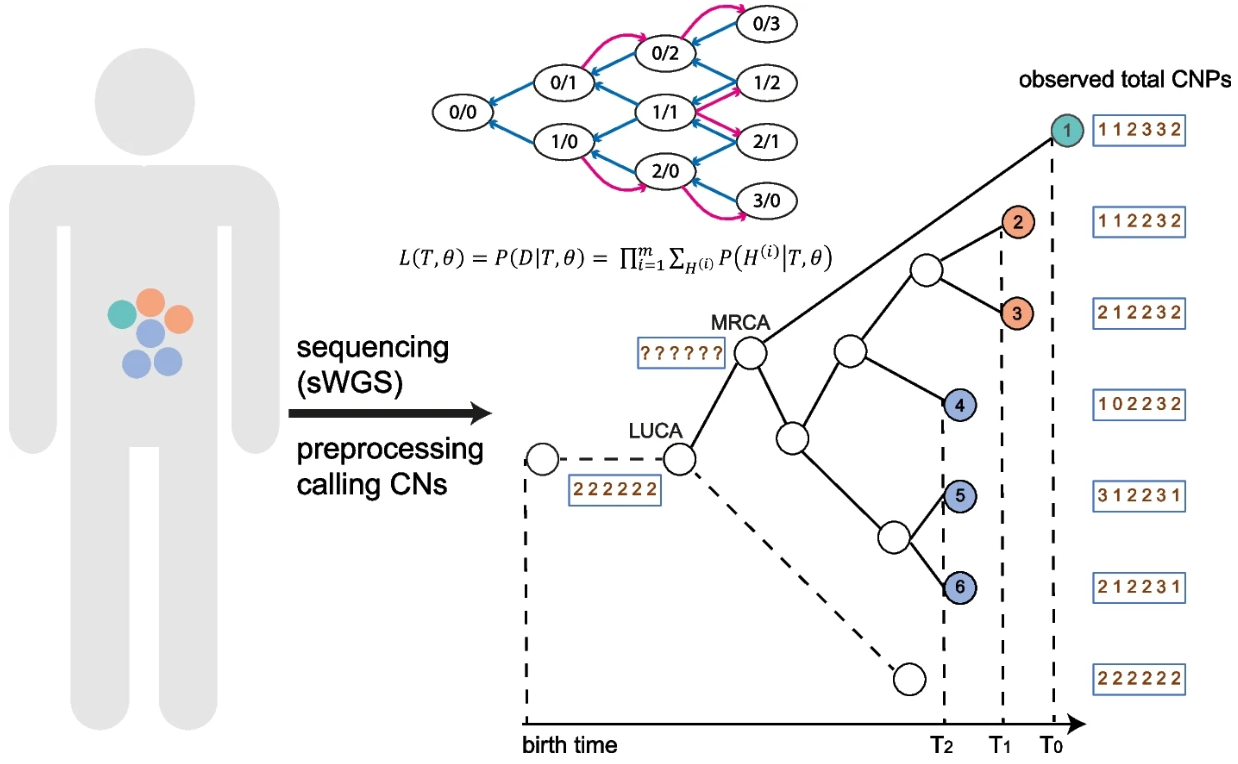
- 在这里,我们介绍 CloMu(Clone To Mutation),一种灵活的低参数癌症进化树生成模型。 CloMu 使用通过强化学习训练的两层神经网络,根据克隆上现有的突变确定新突变的概率。 CloMu 支持多种预测任务,包括确定进化轨迹、树选择、突变之间的因果关系和互换性以及突变适应性。
- 重要的是,以前的方法仅支持其中一些任务,并且许多方法在具有大量突变的数据集上出现过度拟合。通过模拟,我们证明了 CloMu 在各种预测任务上可以匹配或优于当前的方法。特别是,对于具有可互换突变的模拟数据,当前的方法无法像 CloMu 那样有效地揭示因果关系。
- 在乳腺癌和白血病队列中,我们表明 CloMu 可以确定突变之间的相似性和因果关系以及突变的适合度。我们通过将 CloMu 推断的白血病队列突变适合度值与训练期间未使用的克隆比例数据进行比较来验证它们,显示出高度一致性。
- 总之,CloMu 的低参数模型有助于在日益可用的队列级别数据集上执行有关癌症演变的广泛预测任务。
使用 FracMinHash 导出各种进化距离内突变率的置信区间
- 草图方法(Sketching methods)为计算生物学家提供了可扩展的技术来分析不断增长的数据集。 MinHash 是一种估计集合相似性的技术,最近得到了广泛的应用。然而,传统的 MinHash 先前已被证明在应用于大小非常不同的集合时表现不佳。最近引入了 FracMinHash 作为 MinHash 的修改,以弥补集合大小不同时性能的不足。该方法已成功应用于广泛使用的工具 sourmash 中的宏基因组分类学分析。虽然实验证据令人鼓舞,但 FracMinHash 尚未从理论角度进行分析。
- 在本文中,我们进行这样的分析来得出 FracMinHash 的各种统计数据,并证明虽然 FracMinHash 不是无偏的(从某种意义上说,它的期望值不等于它尝试估计的数量),但这种偏差很容易纠正遏制版本和 Jaccard 索引版本。
- 我们展示如何通过假设一个简单的突变模型,使用 FracMinHash 来计算点估计以及一对序列之间进化突变距离的置信区间。我们还调查了这些分析可能失败的边缘情况,以有效地警告 FracMinHash 的用户,表明发生此类情况的可能性。
- 我们的分析表明,与传统 MinHash 相比,FracMinHash 可以更准确、更准确地估计大型宏基因组中基因组的包含情况,并且点估计和置信区间在估计突变距离方面表现明显更好。
快速外显子组测序在近亲人群中的临床应用
The clinical utility of rapid exome sequencing in a consanguineous population. Genome Med
- 背景:外显子组测序的临床实用性现已得到充分记录。快速外显子组测序 (RES) 比常规外显子组测序更需要资源,通常用于特殊的临床环境,其中紧急分子诊断被认为会影响急性治疗。 RES 临床实用性的研究主要限于远交种群。
- 方法:在这里,我们描述了我们在高度近亲人群中进行快速外显子组测序(RES)的经验。临床环境包括重症监护病房、接近法定终止妊娠截止日期的产前病例以及紧急移植决定。
- 结果:189 例病例中有 80 例 (42%) 观察到阳性分子发现(解释表型的致病性或可能致病性变异),而 15 例 (8%) 和 94 例 (50%) 发现不明确(意义不确定的变异) VUS)) 和阴性结果。研究人群的近亲性质使我们有机会观察先前报道的基因的极不寻常和严重的表型表达。在几乎所有(79/80)具有阳性分子发现的病例中观察到临床效用,包括管理决策、预后和生殖咨询。生殖咨询在这一人群中尤其重要,其中绝大多数 (86%) 已识别的变异是常染色体隐性遗传,在这方面,它们比 RES 通常在其他地方报告的从头变异更具可操作性。事实上,我们的成本效益分析表明,研究人群的成本节省令人瞩目。
- 结论:这项工作扩大了 RES 具有明显临床效用的环境的多样性。
跨临床试验的全基因组测序确定了 GPR68 中与化疗引起的周围神经病变相关的罕见编码变异
做了我一直想做的事 (ฅ´ω`ฅ)
- 背景:剂量限制性毒性显着影响许多药物的获益/风险状况。接受具有剂量限制毒性的药物的患者的全基因组测序 (WGS) 可以确定预防这些毒性的治疗假设。化疗引起的周围神经病变 (CIPN) 是一种常见的限制剂量的化疗神经毒性,目前尚无有效的预防方法。
- 方法:我们使用来自 14 项随机对照试验中 4900 名欧洲血统癌症患者的全血样本的 30×种系 WGS 数据,对首次发生周围神经病变事件的时间进行了遗传研究。这些试验中有相当多的患者接受紫杉烷类和铂类化疗作为其治疗方案的一部分,无论是标准治疗还是与 PD-L1 抑制剂 atezolizumab 联合使用。这些试验涉及多种癌症,包括肾细胞癌、三阴性乳腺癌、非小细胞肺癌、小细胞肺癌、膀胱癌、卵巢癌和黑色素瘤。
- 结果:我们在 GRID2 的内含子 13 中确定了一个由低频变异组成的基因座,该基因座与由 rs17020773 索引的第一次周围神经病变 (PN) 的发作时间相关(p = 2.03 × 10-8,所有患者,p = 6.36 × 10-9,紫杉烷处理)。基因水平负荷分析确定了与 GPR68 (一种pH 敏感 G -蛋白偶联受体)的 C 末端罕见编码变异与 PN 风险增加相关(p = 1.59 × 10-6,所有患者,p = 3.47 × 10-8,紫杉烷处理)。发现驱动该信号的变体会改变 GPR68 C 末端预测的抑制蛋白结合基序。对人背根神经节 (DRG) 的 snRNA-seq 分析表明,GPR68 在机械热敏感伤害感受器中表达最高。
- 结论:我们的遗传学研究深入了解了低频和罕见的编码遗传变异对 PN 风险的影响,并表明对感觉神经元中 GPR68 的进一步研究可能会产生预防 CIPN 的治疗假设。
CNETML:从多个样本的拷贝数概况推断系统发育的最大似然
CNETML: maximum likelihood inference of phylogeny from copy number profiles of multiple samples. Genome Biol
- 基于患者多个样本的拷贝数谱的系统发育树有助于了解癌症的进化。
- 在这里,我们开发了一种新的最大似然方法 CNETML,用于从此类数据推断系统发育。 CNETML 是第一个根据纵向样本的总拷贝数联合推断树拓扑、节点年龄和突变率的程序。
- 我们的广泛模拟表明,CNETML 在相对于倍性的拷贝数上表现良好,并且在稍微违反模型假设的情况下表现良好。 CNETML 在实际数据中的应用产生了与之前的发现一致的结果,并为进一步研究提供了新颖的早期拷贝数事件。
方法评估:单细胞组蛋白翻译后修饰
A benchmark of computational pipelines for single-cell histone modification data – PubMed. Genome Biol
- 单细胞组蛋白翻译后修饰 (single-cell histone post translational modification, scHPTM) 检测(例如 scCUT&Tag 或 scChIP-seq)允许对复杂组织内的不同表观基因组景观进行单细胞图谱,并可能解锁我们对发育或疾病中涉及的各种机制的理解。运行 scHTPM 实验并分析产生的数据仍然具有挑战性,因为目前关于实验设计和数据分析流程的良好实践很少存在共识指南。
- 我们执行计算基准来评估实验参数和数据分析流程对细胞表示重现已知生物相似性的能力的影响。我们进行了超过一万次实验,系统地研究了覆盖率和单元数量、计数矩阵构建方法、特征选择和归一化以及所使用的降维算法的影响。这使我们能够确定关键的实验参数和计算选择,以获得单细胞 HPTM 数据的良好表示。
- 我们发现,计数矩阵构建步骤对表示质量有很大影响,并且使用固定大小的 bin 计数优于基于注释的 binning。基于潜在语义索引的降维方法优于其他方法,并且特征选择是有害的,而只要分析足够多的单元,仅保留高质量单元对最终表示影响很小。
- 该基准测试提供了关于实验参数和计算选择如何影响单细胞 HPTM 数据表示的全面研究。我们提出了一系列关于矩阵构造、特征和单元选择以及降维算法的建议。
BCR-ABL1 淋巴细胞白血病的转录组分型
Transcriptomic classes of BCR-ABL1 lymphoblastic leukemia. Nat Genet
转录组分型。一定要了解一下!
- 在 BCR-ABL1 淋巴细胞白血病中,对酪氨酸激酶抑制剂 (TKI) 的治疗异质性知之甚少,尤其是在 BCR-ABL1 不存在激酶结构域突变的情况下。
- 通过深度分子谱分析,我们发现了 BCR-ABL1 淋巴细胞白血病的三种转录组亚型,每一种都代表 B 细胞祖细胞分化阶段的成熟停滞。较早被捕与血统混乱、治疗无效和患者预后不良有关。后来的逮捕与谱系保真度、白血病的持久缓解和患者预后的改善有关。每次成熟停滞都以控制 B 细胞发育中不同转变点的特定基因组事件为标志。
- 有趣的是,无论亚型如何,从患者分离的 BCR-ABL1+ 白血病前干细胞中都不存在这些事件,这支持转录组学表型是在白血病起始事件的下游确定的。
- 总体而言,我们的数据表明治疗反应和 TKI 疗效是这种白血病转化的分化阶段的意外结果。
海马和亚区体积的跨祖先全基因组关联荟萃分析
Cross-ancestry genome-wide association meta-analyses of hippocampal and subfield volumes. Nat Genet
- 海马体对于记忆和认知以及神经精神疾病至关重要,其子区域的结构和功能各不相同。海马体和亚区体积的全基因组关联研究主要在欧洲人群中进行;然而,其他祖先群体的代表性不足。
- 在这里,我们对 65,791 名个体的海马体积和 38,977 名亚区体积进行了跨祖先全基因组关联荟萃分析,其中包括 7,009 名东亚血统个体。我们在 P < 1.13 × 10-9 时确定了 44 个海马特征的 339 个变异特征关联,包括 23 个新关联。
- 尽管存在特定于祖先的关联,但常见的遗传变异对不同祖先的海马特征具有相似的影响。交叉祖先分析提高了精细定位精度和代表性不足人群中多基因分数的预测性能。这些基因变异丰富了 Wnt 信号传导和神经元分化,并影响认知、情绪和神经精神疾病。
- 这些发现可能有助于深入了解海马体和亚区体积的遗传结构。
GWAS定义IgA 肾病的致病信号通路和靶点
堆数据型GWAS
- IgA 肾病 (IgAN) 是一种进行性肾脏疾病,定义为肾小球沉积 IgA。在这里,我们对 17 个国际队列中的 10,146 例肾活检诊断的 IgAN 病例和 28,751 例对照进行了全基因组关联研究。
- 我们定义了 30 个全基因组显着风险位点,解释了 11% 的疾病风险。共有16个新位点,包括TNFSF4/TNFSF18、REL、CD28、PF4V1、LY86、LYN、ANXA3、TNFSF8/TNFSF15、REEP3、ZMIZ1、OVOL1/RELA、ETS1、IGH、IRF8、TNFRSF13B和FCAR。风险位点富含基因直向同源物(gene orthologs, 不同物种中具有相似或相同功能的基因),当对小鼠进行基因操作时,会导致 IgA 水平异常。
- 我们还观察到 IgAN 和血清 IgA 水平之间存在正向遗传相关性。 IgAN 的高多基因评分与早期肾衰竭发病相关。在对候选因果基因的全面功能注释分析中,我们观察到生物候选基因在一组常见的炎症信号通路和细胞因子配体-受体对上的收敛,从而优先考虑潜在的新药物靶点。
TCR-seq的污染检测
A novel statistical method for decontaminating T-cell receptor sequencing data – PubMed. Brief Bioinform
- T 细胞受体 (TCR) 库在人群中高度多样化,在启动多种免疫过程中发挥着重要作用。 TCR 测序 (TCR-seq) 已被开发用于分析 T 细胞库。与其他高通量实验类似,TCR-seq 的几个步骤中可能会发生污染,包括样品收集、制备和测序。这种污染会在数据中产生伪影,导致结果不准确甚至有偏差。大多数现有方法都假设“干净”的 TCR-seq 数据作为起点,无法处理数据污染。
- 在这里,我们开发了一种新颖的统计模型来系统地检测和消除 TCR-seq 数据中的污染。我们将观察到的污染总结为两个来源:成对污染和跨队列污染。对于这两个来源,我们提供可视化和汇总统计数据,以帮助用户评估污染的严重程度。
- 结合来自 14 个现有 TCR-seq 数据集的先验信息,污染最小,我们开发了一个简单的贝叶斯模型来统计识别污染样本。我们进一步提供了删除受影响序列的策略,以便进行下游分析,从而避免重复实验的需要。与模拟研究中的一些现成检测方法相比,我们提出的模型显示了污染检测的鲁棒性。我们说明了我们提出的方法在本地生成的两个 TCR-seq 数据集上的使用。
BBmix:基于RNA测序进行基因分型
BBmix: a Bayesian beta-binomial mixture model for accurate genotyping from RNA-sequencing. Bioinformatics
- 虽然已经开发了许多管道来使用 RNA 测序数据获取基因型,但它们都采用了 DNA 基因型调用器,这些调用器不模拟 RNA 测序特有的偏差,例如等位基因特异性表达。
- 在这里,我们介绍了 BBmix,这是一种贝叶斯 beta-二项式混合模型,它首先学习每种基因型的读取计数的预期分布,然后部署这些学习的参数以概率地调用基因型。
- 我们在各种数据集上对我们的模型进行了基准测试,表明我们的方法通常比竞争对手表现更好,这主要是由于杂合子调用的准确性提高了 1.4%,这可能对降低误报率产生很大影响对基因分型错误敏感的应用,例如等位基因特异性表达。此外,BBmix 可以轻松整合到标准管道中以调用基因型。我们进一步表明,参数通常可以在数据集中转移,因此不到一小时的单次学习运行足以在大量样本中识别基因型。
- 我们将 BBmix 实现为 R 包,可根据 GPL-2 许可在 https://gitlab.com/evigorito/bbmix 和随附的管道 https://gitlab.com/evigorito/bbmix_pipeline 免费获得。
知识图谱嵌入分析转录因子-靶基因相互作用
Knowledge graph embedding for profiling the interaction between transcription factors and their target genes. PLoS Comput Biol
- 转录因子与靶基因之间的相互作用构成了人类基因调控网络的主要部分,这仍然是生物学研究中的复杂因素。具体来说,在已建立的数据库中记录的交互中,有近一半的交互类型尚未得到确认。尽管存在多种计算方法来预测基因相互作用及其类型,但仍然没有可用于仅基于拓扑信息来预测它们的方法。
- 为此,我们在这里提出了一种名为 KGE-TGI 的基于图的预测模型,并在我们专门为此问题构建的知识图上以多任务学习的方式进行训练。 KGE-TGI 模型依赖于拓扑信息,而不是由基因表达数据驱动。
- 在本文中,我们将预测转录因子和目标基因的相互作用类型的任务制定为异构图上链接类型的多标签分类问题,并解决另一个本质上相关的链接预测问题。我们构建了一个地面实况数据集作为基准,并在其上评估了所提出的方法。经过 5 倍交叉实验,该方法在链接预测和链接类型分类任务中的平均 AUC 值分别为 0.9654 和 0.9339。
- 此外,一系列对比实验的结果也证明,知识信息的引入对预测有显着的好处,并且我们的方法在此问题上实现了state-of-the-art的性能。
基于Multiplex-GAM的染色质接触全基因组识别可产生被 Hi-C 忽视的见解
Multiplex-GAM: genome-wide identification of chromatin contacts yields insights overlooked by Hi-C. Nat Methods
- 测量 3D 基因组拓扑的技术对于研究基因调控、基因组组装和基因组重排图谱越来越重要。 Hi-C 和其他基于连接的方法已成为常规方法,但具有特定的偏差。
- 在这里,我们开发了 Multiple-GAM,这是一种更快、更实惠的基因组架构作图 (GAM) 版本,这是一种无需连接的技术,可在全基因组范围内绘制染色质接触图。
- 我们使用小鼠胚胎干细胞对 Multiple-GAM 和 Hi-C 进行了详细比较。当检查这两种方法检测到的最强接触时,我们发现其中只有三分之一是共享的。 GAM 中特别发现的最强接触通常涉及“活性”区域,包括许多转录基因和超级增强子,而在 Hi-C 中,它们通常包含“非活性”区域。
- 我们的工作表明,活跃的基因组区域涉及广泛的复杂接触,而这些接触目前在基于连接的方法中被低估,并强调了全基因组接触图谱技术正交进步的必要性。
欧洲油菜的靶向代谢组学
- 种子含油量是欧洲油菜 (B. napus) 的重要农艺性状,代谢物被认为是物理性状基因型和表型之间的桥梁。
- 通过对 388 个欧洲油菜自交系的自然种群进行广泛靶向的代谢组学分析,我们通过液相色谱质谱法对成熟种子中的 2172 种代谢物进行了量化,其中 131 种标记代谢物被鉴定为与种子油含量相关。然后选择这些代谢物进行进一步的代谢物全基因组关联研究和代谢物转录组范围关联研究。
- 结合加权相关网络分析,我们构建了代谢物、代谢物数量性状位点、基因和共表达模块之间的三重关系网络,该网络包括 21,000 条边和 4384 个节点。我们验证了 BnaA03.TT4、BnaC02.TT4 和 BnaC05.UK 的功能,这三个候选基因通过多组学分析预测,通过调节欧洲油菜中的类黄酮代谢对种子油含量产生显着影响。
- 本研究证明了利用标记代谢物与多组学分析相结合的优势来剖析作物农艺性状的遗传基础。
单细胞组学实验的背景噪音
The effect of background noise and its removal on the analysis of single-cell expression data – PubMed. Genome Biol
- 在基于液滴的单细胞和单核 RNA-seq 实验中,并非所有与一个细胞条形码相关的读数都源自封装的细胞。这种背景噪音归因于无细胞环境 RNA 或条形码交换事件的溢出。
- 我们描述了这种背景噪声的特征,例如小鼠肾脏的三个 scRNA-seq 和两个 snRNA-seq 复制。对于每个实验,汇集来自两个小鼠亚种的细胞,以识别交叉基因型污染分子,从而分析背景噪声。背景噪声在重复和细胞之间变化很大,平均占每个细胞总计数 (UMI) 的 3-35%,我们发现噪声水平与标记基因的特异性和可检测性成正比。在寻找背景噪音的来源时,我们发现了多种证据表明大多数背景分子来自环境 RNA。
- 最后,我们使用基于基因型的估计来评估旨在量化和消除背景噪声的三种方法(CellBender、DecontX、SoupX)的性能。我们发现 CellBender 提供了最精确的背景噪声水平估计,并且对标记基因检测也产生了最高的改进。相比之下,细胞的聚类和分类对于背景噪声相当稳健,并且通过背景去除只能实现很小的改进,这可能会以精细结构的扭曲为代价。
- 我们的研究结果有助于更好地了解单细胞实验中背景噪声的范围、来源和影响,并为如何处理它提供指导。
综述类
神经球全基因组汇集 CRISPR 筛选
Genome-wide pooled CRISPR screening in neurospheres. Nat Protoc
- 球体培养系统允许无法在标准细胞培养条件下生长的细胞进行体外繁殖,并且可以比当前模型系统更好地捕获模拟肿瘤生长的细胞环境。从对传统培养条件下生长的数千个癌细胞系进行全基因组聚类规则间隔短回文重复 (CRISPR) 筛选中收集到的见解说明了此类 CRISPR 合并筛选的价值。显然,三维球体培养物的类似全基因组 CRISPR 筛选对于未来的生物发现非常重要。
- 在这里,我们提出了三维神经球全基因组 CRISPR 筛选的方案。虽然针对更典型的细胞系已经发表了许多深入的方案和讨论,但目前文献中很少有可用于球状细胞系全基因组筛选的详细方案。对于那些想要筛选此类细胞系,特别是神经球的人,我们提供了筛选前要进行的分析开发测试以及筛选本身的分步说明。我们强调了对变量的考虑,这些变量使这些筛选不同于或类似于典型的非球状细胞系。最后,我们说明了神经球全基因组筛选的典型结果,以及神经球筛选通常如何产生比更多典型癌细胞系稍微更异质的信号分布。从最初的分析开发测试到测序数据的反卷积,整个协议的完成将需要 8-12 周的时间。
FDA获批的肿瘤治疗产品
Trends in the approval of cancer therapies by the FDA in the twenty-first century. Nat Rev Drug Discov
- 自世纪之交以来,癌症治疗格局发生了巨大变化,患者的治疗结果得到了显着改善。本综述总结了 FDA 从 2000 年 1 月到 2022 年 10 月期间批准肿瘤治疗产品的趋势,根据这些产品的作用机制和主要靶点进行分类。
- 值得注意的是,在靶向治疗批准的推动下,这段时间肿瘤适应症的批准率有所增加,新治疗方法的引入率也有所增加。从批准的产品数量和适应症来看,激酶抑制剂是主导产品类别,而免疫检查点抑制剂尽管直到 2011 年才进入市场,但批准数量位居第二。其他趋势包括生物标志物定义人群的批准比例略有增加,以及与肿瘤部位无关的批准的出现。最后,我们考虑趋势对肿瘤治疗产品开发未来的影响,包括新型治疗方法和技术的影响。
基于单细胞组学的肿瘤内皮细胞异质性研究
Understanding tumour endothelial cell heterogeneity and function from single-cell omics . Nat Rev Cancer
- 抗血管生成疗法(AAT)用于治疗不同类型的癌症。然而,由于功效和抵抗力不足,它们的成功有限。最近,肿瘤内皮细胞(TEC)的单细胞组学研究提供了新的机制见解。
- 在这里,我们在单细胞水平上概述了迄今为止研究的所有肿瘤类型的人类 TEC 的异质性。值得注意的是,大多数人类肿瘤类型含有不同数量但只有一小部分的血管生成 TEC(AAT 的假定靶标),这可能导致 AAT 的疗效有限和耐药性有限。一般来说,所有肿瘤类型内部和跨肿瘤类型的 TEC 都是异质的,但由于缺乏统一的内皮细胞命名法和一致的单细胞分析方案,比较肿瘤之间的 TEC 表型目前具有挑战性,迫切需要更一致的方法方法。尽管如此,在大多数肿瘤类型中,可以识别通用 TEC 标记(ACKR1、PLVAP 和 IGFBP3)。除了血管生成之外,免疫调节和细胞外基质组织等生物过程也是不同肿瘤类型中最常预测的 TEC 富集特征之一。
- 尽管血管生成和细胞外基质靶标已被考虑用于 AAT(未取得预期的成功),但 TEC 的免疫调节特性尚未被充分考虑为一种新型抗癌治疗方法。
- 因此,我们还讨论了 AAT 发展的进展、局限性、解决方案和新目标。
eMERGE网络的临床转化挑战
Studying the impact of translational genomic research: Lessons from eMERGE. Am J Hum Genet
- 电子病历和基因组学 (the Electronic Medical Record and Genomics, eMERGE) 网络的两个主要目标是了解如何最好地将研究结果返回给患者/参与者以及照顾他们的临床医生,并评估将这些结果应用于临床护理的影响。然而,自成立以来,该网络在实现这些目标方面面临着一系列挑战,其中许多挑战具有需要考虑的道德、法律或社会影响 (ethical, legal, or social implications, ELSI)。
- 在这里,我们分享了我们在招募参与者、返回结果和评估其影响方面遇到的障碍,所有这些都影响了我们实现 eMERGE 目标的能力,以及我们为尝试解决这些障碍而采取的步骤。
- 我们将遇到挑战的领域分为四大类:(1)研究设计,包括招募更多样化的群体; (2) 同意; (3) 将结果返回给参与者及其医疗保健提供者 (HCP); (4) 评估参与者的后续护理并衡量研究对参与者及其家人的影响。由于 eMERGE 的大多数阶段都包括儿童和成人,因此我们还通过将儿童群体纳入本研究来解决特殊的 ELSI。
- 我们为改进转化基因组研究提出具体建议,以确保未来的项目能够有效地返回结果并评估其对患者/参与者和提供者的影响,如果要实现基因组信息医学的目标。
流行病学类
人类乳头瘤病毒与美国口咽癌发病率上升
Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J Clin Oncol
这结果怎么透露着一种古怪 Σ( ° △ °|||)︴
- 目的:最近美国口咽癌发病率和生存率的增加被归因于人乳头瘤病毒(HPV)感染,但缺乏经验证据。
- 患者和方法:通过使用聚合酶链反应和基因分型,确定了 SEER 数据库中三个基于人群的癌症登记处收集的所有 271 例口咽癌 (1984-2004) 的 HPV 状态(Inno-LiPA)、HPV16 病毒载量和 HPV16 mRNA 表达。使用逻辑回归估计四个日历期间 HPV 患病率的趋势。将观察到的 HPV 患病率重新加权到癌症登记处的所有口咽癌中,以考虑非随机选择并计算发病率趋势。使用 Kaplan-Meier 和多变量 Cox 回归分析比较 HPV 阳性和 HPV 阴性患者的生存率。
- 结果:无论 HPV 检测结果如何,口咽癌中 HPV 的患病率随日历时间显着增加(P 趋势 < .05)。例如,Inno-LiPA 使 HPV 患病率从 1984 年至 1989 年期间的 16.3% 上升至 2000 年至 2004 年期间的 71.7%。HPV 阳性患者的中位生存期明显长于 HPV 阴性患者(131 个月与 20 个月;对数秩 P < .001;调整后的风险比,0.31;95% CI,0.21 至 0.46)。 HPV 阳性患者的存活率在整个日历周期内显着增加 (P = .003),但 HPV 阴性患者的存活率则没有显着增加 (P = .18)。从 1988 年到 2004 年,人群中 HPV 阳性口咽癌的发病率增加了 225%(95% CI,208% 至 242%)(从每 100,000 人 0.8 例增加到每 100,000 人 2.6 例),而 HPV 阴性癌症的发病率下降了 50% %(95% CI,47% 至 53%;从每 100,000 人 2.0 到每 100,000 人 1.0)。如果最近的发病率趋势持续下去,预计到 2020 年,每年 HPV 阳性口咽癌的数量将超过每年宫颈癌的数量。
- 结论:自 1984 年以来,美国口咽癌人口发病率和生存率的增加是由 HPV 感染引起的。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍
