本博客由科研AI Agent实验室BenszResearch强力驱动!如何更快地访问本站?有需要可加电报群获得更多帮助。本博客用什么VPS?创作不易,请支持苯苯!推荐购买本博客的VIP喔,10元/年即可畅享所有VIP专属内容!
概览
- 连接进化、生理学和机器人物理学中的两种昆虫飞行模式
- 唾液酸聚糖结合触发人类冠状病毒中的刺突开放
- PTPN2/PTPN1 抑制剂 ABBV-CLS-…
- 英国生物库中血浆蛋白质组与遗传学和健康的关联
- 英国生物银行中罕见变异与血浆蛋白水平的关联
前言
本文是前沿快讯的第41期。前沿快讯栏目主要收集一些个人感兴趣的近期发表的研究,关注领域包括肿瘤的分子生物学、临床研究、流行病学等,文献类型主要是期刊论文和综述。研究介绍在Google机翻摘要的基础上进行微调,可能不一定特别准确、专业,主要目的是方便自己和大家快速了解和回顾相关领域研究进展。如果你对某个研究的细节感兴趣,请自行寻找全文进一步了解。此外,研究根据子领域会进一步细分,不过交叉领域的研究不好分为某一类,所以这个分类主要用于初级索引,并不十分准确,不喜勿喷。最后,大家看到什么特别的研究,也可以在评论区向我推荐,我会酌情收录在后面的期刊中。如无意外,前沿快讯栏目会长期更新,周期为2周-1月不等。从第5期开始,前沿快讯会新增一个CNS类,用来记录一些发表在Nature, Science或Cell杂志上的研究。从第18期开始,“肿瘤转移类”、“肿瘤代谢类”等将不再更新,而是合并至其它分类。
本期有以下知识点值得关注:
CNS类
连接进化、生理学和机器人物理学中的两种昆虫飞行模式
Bridging two insect flight modes in evolution, physiology and robophysics. Nature
- 自从飞行以来,昆虫在两种看似不同的飞行模式之间经历了反复的进化转变。一些昆虫的神经系统会在每次拍翅时同步激活它们的肌肉。然而,许多昆虫通过进化与神经激活异步并响应机械拉伸而激活的飞行肌肉,已经达到了超出典型神经肌肉系统速度限制的翅膀拍动频率。这些模式反映了产生节奏运动的两种基本方式:时间周期强迫与自激产生的突发振荡。重复的进化转变是如何发生的以及是什么控制着这些不同模式之间的切换仍然未知。
- 在这里,我们发现,尽管昆虫在整个系统发育过程中存在广泛的异步驱动,但异步可能只在目级进化过一次,并多次回归到祖先的同步模式。从异步祖先进化而来的同步蛾物种仍然保留了拉伸激活的肌肉生理机能。对统一生物物理框架的数值和机器人物理分析表明,这两种模式不是二分法,而是同一动力学的两种状态。昆虫可以通过生理参数空间中的桥梁在飞行模式之间转换。
- 最后,我们将这两种驱动模式集成到昆虫规模的机器人中,该机器人能够在模式之间进行转换,并为工程飞行解锁新的自激翼泳策略。
- 总之,该框架解释了昆虫飞行进化中的重复转变,并展示了飞行模式如何随着生理参数的变化而翻转。
唾液酸聚糖结合触发人类冠状病毒中的刺突开放
Sialoglycan binding triggers spike opening in a human coronavirus. Nature
- 冠状病毒刺突蛋白介导受体结合和膜融合,使其成为中和抗体的主要目标。在严重急性呼吸综合征冠状病毒、严重急性呼吸综合征冠状病毒 2 型和中东呼吸综合征冠状病毒中,刺突蛋白在开放和闭合构象之间自由转换,以平衡宿主细胞附着和免疫逃避。刺突开放暴露了结构域 S1B,使其能够与蛋白质受体结合,并且还被认为能够在膜融合过程中实现蛋白质重折叠。然而,除了一个例外,迄今为止研究的所有其他冠状病毒的融合前刺突蛋白都是在闭合状态下观察到的。这增加了调节的可能性,刺突蛋白更常见地响应特定的线索而不是自发地转变为开放状态。
- 在这里,利用低温电子显微镜和分子动力学模拟,我们发现普通感冒人类冠状病毒 HKU1 的刺突蛋白在将基于唾液酸聚糖的初级受体与结构域 S1A 结合后发生局部和远程构象变化。这种结合通过变构域间串扰触发 S1B 域向开放状态的转变。我们的研究结果提供了对冠状病毒附着的详细了解,以及使用双受体和启动进入作为免疫逃逸手段的可能性。
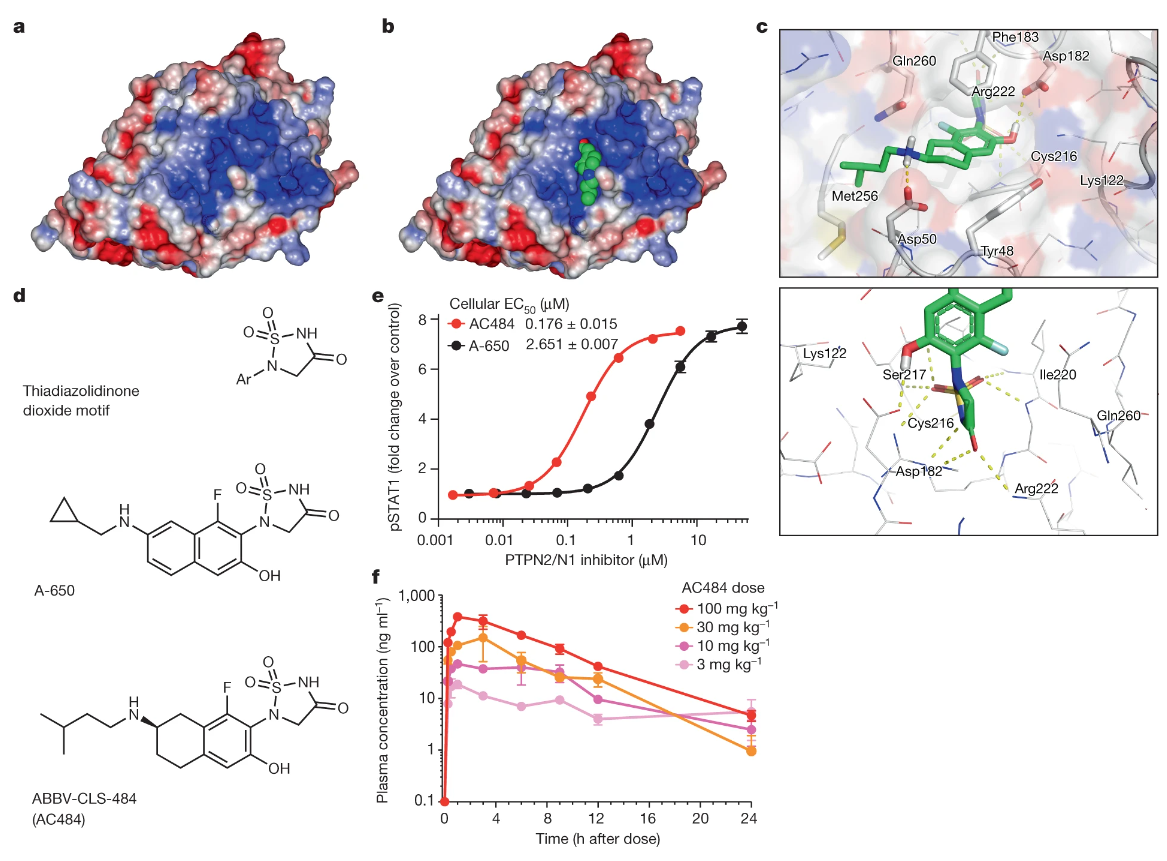
PTPN2/PTPN1 抑制剂 ABBV-CLS-484 释放强大的抗肿瘤免疫力
The PTPN2/PTPN1 inhibitor ABBV-CLS-484 unleashes potent anti-tumour immunity. Nature
我以前一直以为磷酸酶抑制剂类药物在临床上是比较常见 Σ( ° △ °|||)︴
- 免疫检查点阻断对某些癌症患者有效,但大多数患者对当前的免疫疗法难以治疗,需要新的方法来克服耐药性。蛋白酪氨酸磷酸酶 PTPN2 和 PTPN1 是炎症的核心调节因子,它们在肿瘤细胞或免疫细胞中的基因缺失可促进抗肿瘤免疫。然而,磷酸酶是具有挑战性的药物靶点;特别是,活性位点被认为是不可药物的。
- 在这里,我们介绍 ABBV-CLS-484 (AC484) 的发现和表征,这是一种一流的、口服生物可利用的、有效的 PTPN2 和 PTPN1 活性位点抑制剂。 AC484 体外治疗可增强对干扰素的反应,并促进多种免疫细胞亚群的激活和功能。在对 PD-1 阻断具有抗性的癌症小鼠模型中,AC484 单一疗法可产生有效的抗肿瘤免疫力。我们发现,AC484 通过增强 JAK-STAT 信号传导和减少 T 细胞功能障碍,使肿瘤微环境发炎,并促进自然杀伤细胞和 CD8+ T 细胞功能。 PTPN2 和 PTPN1 抑制剂为癌症免疫治疗提供了一种有前景的新策略,目前正在晚期实体瘤患者中进行评估(ClinicalTrials.gov 标识符 NCT04777994)。
- 更广泛地说,我们的研究表明,关键细胞内免疫调节剂的小分子抑制剂可以在临床前模型中实现与基于抗体的免疫检查点阻断相当或超过的功效。最后,据我们所知,AC484 是第一个进入癌症免疫治疗临床评估的活性位点磷酸酶抑制剂,并可能为针对这一类重要酶的其他治疗方法铺平道路。
- Bensz解读:基本策略是基于结构的药物设计。二氧化噻二唑烷酮等结构基序已经被发现是有效的,因此作者就在其结构基础上进行微调。大致过程:二氧化噻二唑烷酮 $\Rightarrow$ A-650(优化了二氧化噻二唑烷酮环和萘基侧链附近的相互作用)$\Rightarrow$ AC484(萘基部分饱和以获得相应的氨基四氢化萘并优化胺侧链)
英国生物库中血浆蛋白质组与遗传学和健康的关联
Plasma proteomic associations with genetics and health in the UK Biobank. Nature
系列研究
- 制药蛋白质组学项目是一个竞争前生物制药联盟,旨在描述 54,219 名英国生物银行参与者的血浆蛋白质组学特征。在这里,我们提供了该计划的详细总结,包括技术和生物学验证、对蛋白质组疾病特征的见解以及各种人口和健康指标的预测模型。我们展示了 2,923 个蛋白质的全面蛋白质数量性状位点 (pQTL) 作图,识别了 14,287 个主要遗传关联,其中 81% 以前未被描述,同时还提供了非欧洲个体的祖先特异性 pQTL 作图。该研究提供了血浆蛋白质组遗传结构的最新表征,并结合了随着样本量和蛋白质组检测覆盖范围随着时间的推移而增加的预计 pQTL 发现率。我们提供跨多个生物领域的反式 pQTL 的广泛见解,强调遗传对配体-受体相互作用的影响以及跨多种细胞因子和补体网络的通路扰动,并说明 ABO 血型和 FUT2 分泌状态对蛋白质的长期上位效应具有胃肠道组织富集表达。我们通过扩展 PCSK9 等蛋白质靶点对其他终点的遗传代理效应,并解开与 COVID-19 易感性相关的位点受到干扰的特定基因和蛋白质,证明了这些数据在药物发现中的实用性。这种公私合作伙伴关系为科学界提供了相当广度和深度的开放获取蛋白质组学资源,有助于阐明蛋白质基因组发现背后的生物学机制,并加速生物标志物、预测模型和治疗方法的开发1。
英国生物银行中罕见变异与血浆蛋白水平的关联
Rare variant associations with plasma protein levels in the UK Biobank. Nature
系列研究
- 整合人类基因组学和蛋白质组学可以帮助阐明疾病机制、识别临床生物标志物并发现药物靶点。由于之前的蛋白质组学研究主要通过全基因组关联研究来关注常见变异,因此稀有变异对血浆蛋白质组的贡献在很大程度上仍然未知。
- 在这里,我们确定了罕见蛋白质编码变异与在 49,736 名英国生物银行个体中测量的 2,923 个血浆蛋白丰度之间的关联。我们的变异水平外显子组范围关联研究确定了 5,433 个罕见的基因型-蛋白质关联,其中 81% 在之前同一队列的全基因组关联研究中未检测到。然后,我们使用基因水平折叠分析来观察聚合信号,结果揭示了 1,962 个基因-蛋白质关联。在来自蛋白质截短变体的 691 个基因水平信号中,99.4% 与蛋白质水平降低相关。 STAB1 和 STAB2 编码参与血浆蛋白清除的清道夫受体,作为多效性基因座出现,分别与 77 个和 41 个蛋白关联。
- 我们通过多个应用程序展示了可公开访问的资源的实用性。其中包括详细说明 NLRC4 中的等位基因系列,识别 HSD17B13 中脂肪肝疾病相关变异的潜在生物标志物,以及通过在折叠分析中整合蛋白质数量性状基因座与蛋白质截短变异来支持全表型关联研究。最后,我们发现了克隆造血 (CH) 的独特蛋白质组学后果,包括 TET2-CH 与 FLT3 水平升高之间的关联。
- 我们的结果强调了血浆蛋白丰度的罕见变化的重要作用以及蛋白质基因组学在治疗发现中的价值。
通过遗传学和疾病关联进行大规模血浆蛋白质组学比较
Large-scale plasma proteomics comparisons through genetics and disease associations. Nature
系列研究
- 高通量蛋白质组学平台可测量血浆中数千种蛋白质,并结合基因组和表型信息,有能力弥合基因组和疾病之间的差距。在这里,我们对英国生物银行制药蛋白质组学项目生成的 Olink Explore 3072 数据进行了关联研究,这些数据对来自 50,000 多名英国生物银行参与者的血浆样本、表型和基因型数据进行了关联研究,对英国或爱尔兰、非洲和南亚血统进行了分层。我们将结果与 SomaScan v4 对 36,000 名冰岛人血浆进行的研究进行了比较,其中 1,514 人的 Olink 数据也可用。
- 我们发现两个平台之间存在一定的相关性。尽管在两个平台上检测到的顺式蛋白数量性状位点的绝对数量相似(Olink 上为 2,101,SomaScan 为 2,120),但具有此类分析性能支持证据的检测比例在 Olink 平台上更高(Olink 为 72%,SomaScan 为 72%)。 43%)。相当多的蛋白质在平台之间具有不同的基因组关联。我们提供了一些例子,其中平台之间的差异可能会影响蛋白质水平与疾病研究相结合得出的结论。
- 我们展示了如何利用英国生物银行参与者的不同血统帮助检测新的关联并完善基因组位置。我们的结果显示了两个最常用的高通量蛋白质组学平台提供的信息的价值,并证明了它们之间的差异,有时提供了有用的互补性。
(~ ̄▽ ̄)~ 组装理论解释并量化了选择和进化
Assembly theory explains and quantifies selection and evolution. Nature. full html. full pdf
英国格拉斯哥大学化学学院
很有趣的研究视角和理论。AT也是基于最小作用量原理的一种理论。
- 科学家们一直在努力协调生物进化与物理学定义的宇宙不变定律。这些定律支撑着生命的起源、进化以及人类文化和技术的发展,但它们并不能预测这些现象的出现。进化论通过选择的视角解释了为什么有些事物存在而另一些事物不存在。为了理解在没有固有设计蓝图的情况下物理学如何产生多样化的、开放式的形式,需要一种理解和量化选择的新方法。
- 我们将组装理论(AT)作为一个框架,它不会改变物理定律,但会重新定义这些定律所作用的“对象”的概念。 AT 将物体概念化为由其可能的形成历史定义的实体,而不是点粒子。这允许对象在个人或选定单位的明确边界内显示选择的证据。我们引入了一种称为组装 (A) 的度量,捕获生成给定对象集合所需的因果关系程度。
- 这种方法使我们能够将新颖性的生成和选择融入到复杂物体的物理学中。它解释了如何通过考虑其组装的正向动态过程来表征这些物体。通过重新构想装配空间中的物质概念,AT 在物理学和生物学之间提供了一个强大的接口。它揭示了在化学尺度上出现的物理学的一个新方面,历史和因果偶然性影响着现存的事物。
血红蛋白体在软骨细胞缺氧适应中的红细胞外作用
An extra-erythrocyte role of haemoglobin body in chondrocyte hypoxia adaption. Nature
第四军医大学空军医学中心基础医学院和西京医院肿瘤生物学国家重点实验室病理科
临床上哪些病理状态会导致 Hedy 损失?
- 尽管血红蛋白是红细胞中已知的氧载体,具有远距离运输氧气的功能,但血红蛋白在红细胞外的生理作用在很大程度上是难以捉摸的。
- 在这里,我们发现软骨细胞产生大量的血红蛋白,在其细胞质中形成伊红阳性小体。血红蛋白体(Hedy)是一种无膜冷凝物,其特征是相分离。软骨细胞中血红蛋白的产生受缺氧控制,并且依赖于 KLF1 而不是 HIF1/2α 途径。
- 软骨细胞中血红蛋白的缺失导致 Hedy 损失,并伴有严重缺氧、糖酵解增强和软骨组织中心广泛的细胞死亡,这是由于缺氧条件下 Hedy 控制的氧供应丧失所致。
- 这些结果证明了血红蛋白在软骨细胞中的红细胞外作用,并揭示了软骨细胞通过 Hedy 在缺氧环境中生存的迄今为止未被认识的机制。
表观遗传平衡确保 MLL 扩增和重排的机械控制
Epigenetic balance ensures mechanistic control of MLL amplification and rearrangement. Cell
- MLL/KMT2A 扩增和易位在婴儿、成人和治疗诱发的白血病中普遍存在。然而,这些改变的分子贡献者尚不清楚。
- 在这里,我们证明 MLL/KMT2A 基因座的组蛋白 H3 赖氨酸 9 单甲基化和二甲基化 (H3K9me1/2) 平衡调节这些扩增和重排。这种平衡由赖氨酸脱甲基酶 KDM3B 和甲基转移酶 G9a/EHMT2 之间的串扰控制。 KDM3B 耗尽会增加 H3K9me1/2 水平并减少 MLL/KMT2A 基因座上的 CTCF 占用,进而促进扩增和重排。 CTCF 的消耗也足以产生这些病灶改变。此外,化疗药物阿霉素 (Dox) 与治疗引起的白血病相关,促进 MLL/KMT2A 扩增和重排,抑制 KDM3B 和 CTCF 蛋白水平。 KDM3B 和 CTCF 过表达可挽救 Dox 诱导的 MLL/KMT2A 改变。人类细胞或小鼠中的 G9a 抑制也会抑制伴随 Dox 治疗的 MLL/KMT2A 事件。
- 因此,MLL/KMT2A 扩增和重排是由表观遗传调节因子控制的,这些调节因子是易于处理的药物靶点,具有临床意义。
无序区域通过凝结和伙伴招募来控制 cBAF 活性
A disordered region controls cBAF activity via condensation and partner recruitment. Cell
看不懂
- 本质无序区域(IDR)占核蛋白总含量的很大一部分。普遍的教条是 IDR 参与非特异性相互作用,因为它们很少受到进化选择的限制。
- 在这里,我们证明了冷凝物形成和异型相互作用是 mSWI/SNF 染色质重塑器 cBAF 的 ARID1A/B 亚基内 IDR 的独特且可分离的特征,并在每个贡献的基础上建立了不同的“序列语法”。缩合是由均匀分布的酪氨酸残基驱动的,伙伴相互作用是由富含丙氨酸、甘氨酸和谷氨酰胺残基的非随机块介导的。
- 这些特征集中了特定的 cBAF 蛋白质-蛋白质相互作用网络,对于染色质定位和活性至关重要。重要的是,ARID1B IDR 序列语法中与人类疾病相关的扰动会破坏细胞中的 cBAF 功能。
- 这些数据共同确定了 IDR 对染色质重塑的贡献,并解释了相分离如何提供一种机制,通过该机制实现基因组定位和功能伙伴招募。
减少临床 CRISPR-Cas9 工程 T 细胞中的染色体丢失
Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell
这个工艺具体是指什么?
- CRISPR-Cas9 基因组编辑使先进的 T 细胞疗法成为可能,但目标染色体的偶尔丢失仍然是一个安全问题。为了调查 Cas9 诱导的染色体丢失是否是一种普遍现象并评估其临床意义,我们对原代人类 T 细胞进行了系统分析。
- 阵列和汇集的 CRISPR 筛选显示,染色体丢失在整个基因组中普遍存在,并导致目标染色体部分和全部丢失,包括在临床前嵌合抗原受体 T 细胞中。染色体丢失的 T 细胞在培养物中持续存在数周,这意味着可能会干扰临床使用。
- 我们首次对 Cas9 工程化 T 细胞 (NCT03399448) 进行人体临床试验时采用了改良的细胞制造工艺,减少了染色体丢失,同时在很大程度上保留了基因组编辑功效。 p53 的表达与本方案中观察到的染色体丢失保护相关,这表明 T 细胞工程的机制和策略可以减轻临床中的这种遗传毒性。
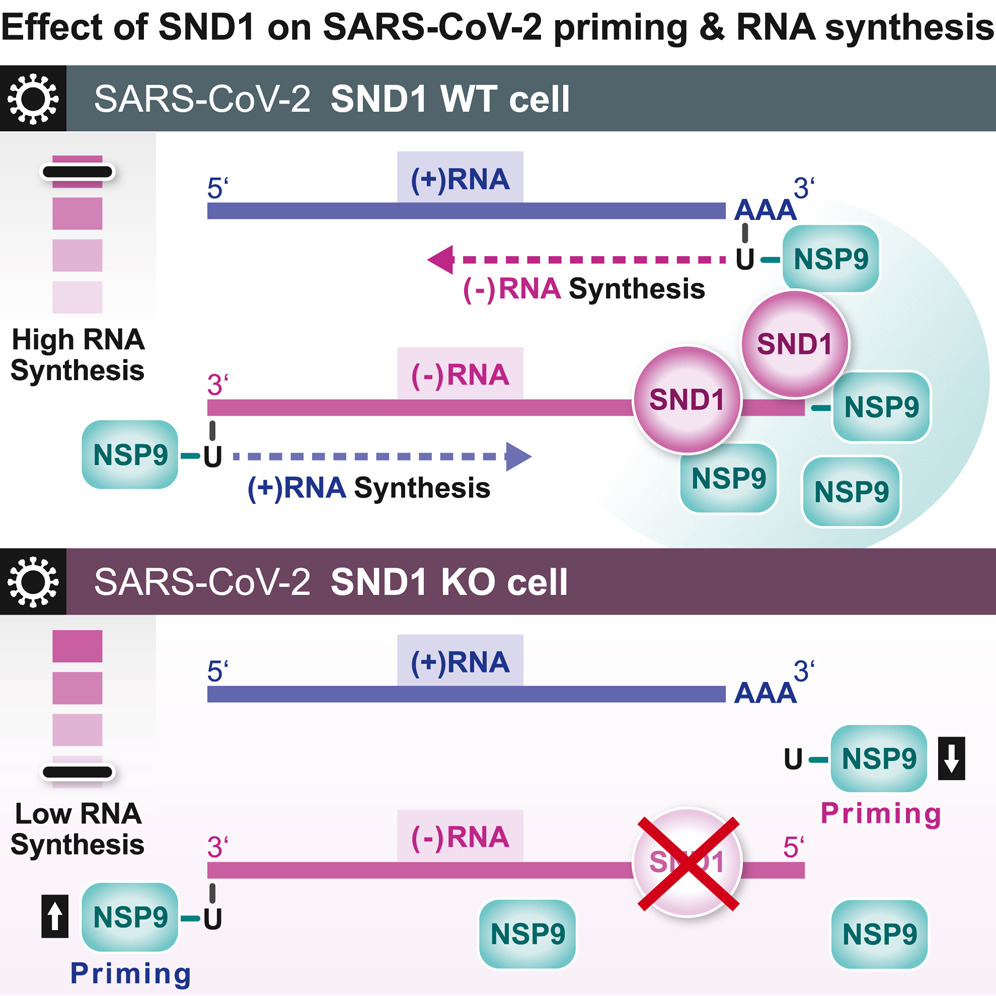
SND1结合SARS-CoV-2负义RNA并通过NSP9促进病毒RNA合成
SND1 binds SARS-CoV-2 negative-sense RNA and promotes viral RNA synthesis through NSP9. Cell
- 病毒 RNA 生物发生的调节是有效 SARS-CoV-2 感染的基础。为了表征这一过程中涉及的宿主 RNA 结合蛋白 (RBP),我们通过生化方法鉴定了与基因组和亚基因组 SARS-CoV-2 RNA 结合的蛋白质。
- 我们发现宿主蛋白 SND1 结合负义病毒 RNA 的 5′ 端,并且是 SARS-CoV-2 RNA 合成所必需的。 SND1耗尽的细胞形成较小的复制细胞器并表现出病毒生长动力学减弱。我们发现 NSP9(一种病毒 RBP 和直接的 SND1 相互作用伴侣)与感染过程中产生的正义和负义 RNA 的 5′ 端共价连接。这些连接发生在复制转录起始位点,与 NSP9 启动病毒 RNA 合成一致。
- 从机制上讲,SND1 重塑 NSP9 占据并改变 NSP9 与病毒 RNA 中起始核苷酸的共价连接。
- 我们的研究结果表明 NSP9 参与了 SARS-CoV-2 RNA 合成的启动,并揭示了细胞蛋白在协调病毒 RNA 生产中的意想不到的作用。
胆碱能神经元触发上皮 Ca2+ 电流维持果蝇中肠稳态
Cholinergic neurons trigger epithelial Ca2+ currents to heal the gut . Nature
- 再生生物学中一个尚未解决的基本问题是组织在受伤后如何恢复稳态。回答这个问题对于了解炎症性肠病和癌症等慢性疾病的病因至关重要。
- 我们使用果蝇中肠对此进行了研究,发现在再生过程中,胆碱能3神经元亚群会触发肠上皮细胞(肠上皮细胞)之间的Ca2+电流,以促进恢复稳态。我们发现,肠道上皮中保守的胆碱能酶乙酰胆碱酯酶的下调使得来自特定TNF/Egr5感应胆碱能神经元的乙酰胆碱能够激活受神经支配的肠细胞中的烟碱受体。这种激活触发高 Ca2+ 通过 Inx2/Inx7 间隙连接在上皮细胞中扩散,促进肠上皮细胞成熟,从而减少增殖和炎症。破坏这一过程会导致慢性损伤,包括离子失衡、Yki/Yap 激活7、细胞死亡和炎症细胞因子增加(让人想起炎症性肠病)。
- 总而言之,保守的胆碱能通路促进上皮 Ca2+ 电流,从而治愈肠上皮。我们的研究结果证明了神经和生物电依赖性肠道再生,并增进了我们目前对组织在受伤后如何恢复稳态的理解。
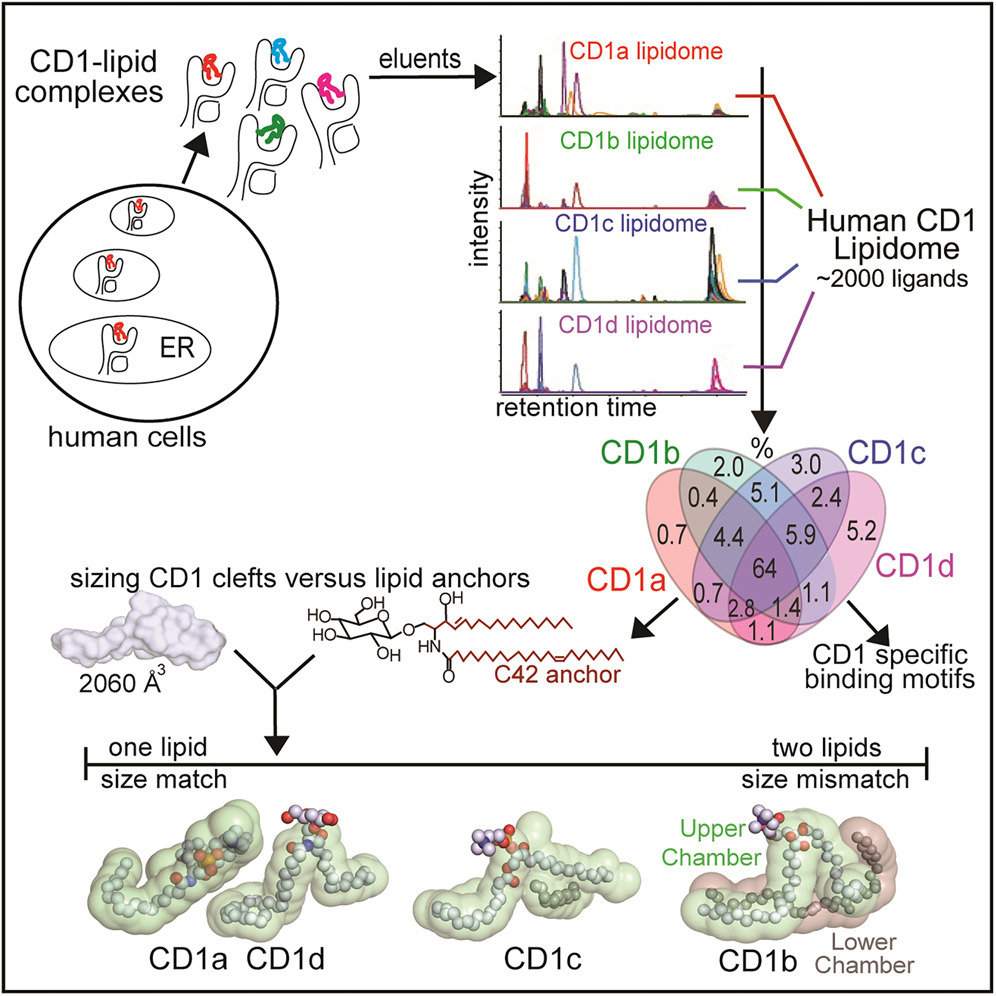
CD1脂质组揭示脂质结合基序和基于大小的抗原展示机制
CD1 lipidomes reveal lipid-binding motifs and size-based antigen-display mechanisms. Cell
- CD1 系统结合脂质抗原以展示给 T 细胞。在这里,我们解析了四种人类 CD1 抗原呈递分子的脂质组,提供了自脂质展示图。>2,000 个 CD1-脂质复合物的检测表明自鞘脂和磷脂的广泛存在。虽然肽抗原是经过化学处理的,但许多脂质以未改变的形式存在。
- 然而,每种类型的 CD1 蛋白都会对自身脂质组进行差异性编辑,以根据脂质长度和化学成分显示不同的捕获基序,这表明了一般的抗原展示机制。对于 CD1a 和 CD1d,脂质大小与 CD1 裂隙体积相匹配。 CD1c 裂隙大小变化较大,而 CD1b 是异常值,其中配体和裂隙显示出极端的大小不匹配,这是通过在一个裂隙中均匀分布两个小脂质来解释的。
- 此外,包含整合 CD1 脂质组的化合物列表支持了脂质阻滞剂和 T 细胞抗原的不断发现。
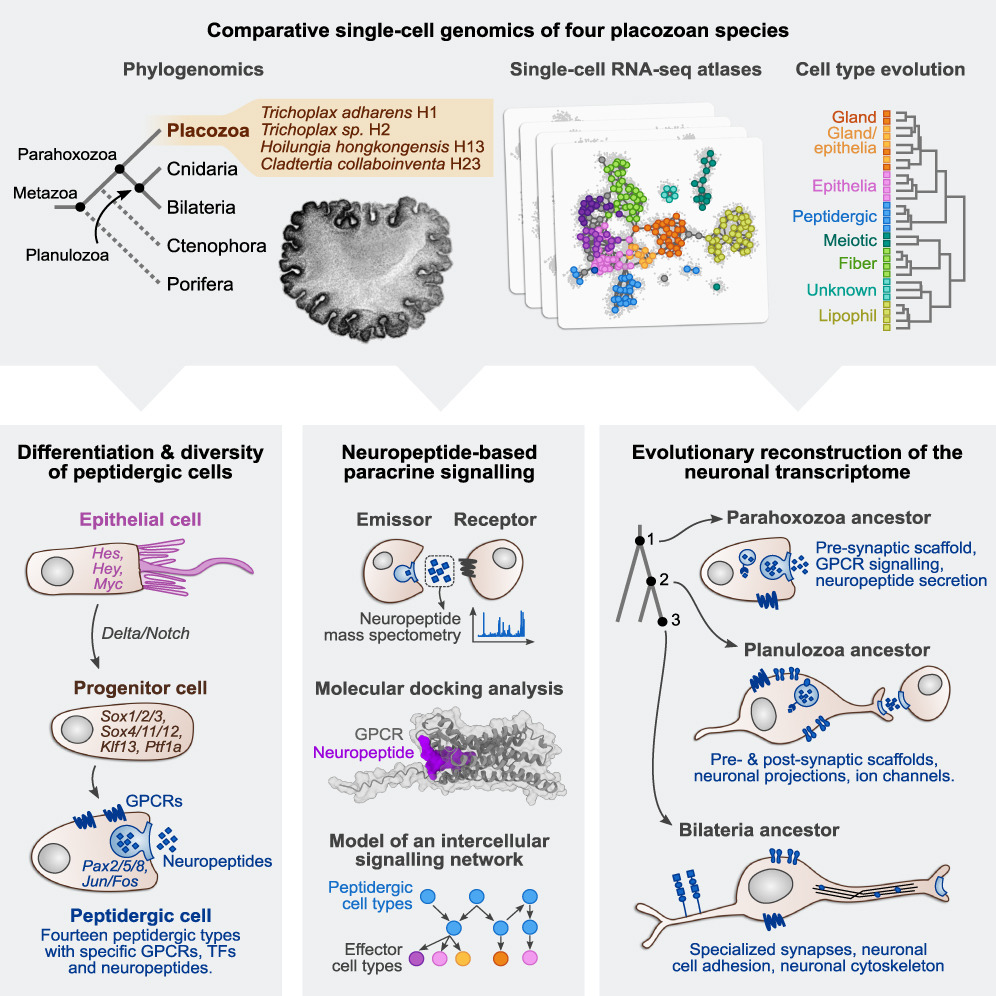
早期动物进化中神经元基因表达程序的逐步出现
Stepwise emergence of the neuronal gene expression program in early animal evolution. Cell
- 神经元和其他主要细胞类型程序的组装发生在动物进化的早期。我们可以通过研究长生动物等非两侧对称动物来重建这个过程。这些小型圆盘状动物不仅具有九种形态学上描述的细胞类型并且没有神经元,而且还表现出由肽分泌细胞触发的协调行为。我们利用四种长生动物的系统发育学、染色质分析和比较单细胞基因组学研究了这些肽能细胞可能的神经元亲和力。
- 我们发现长生动物中保守的细胞类型表达程序,包括转分化和循环细胞群,表明活跃的细胞类型稳态。我们还发现了十四种表达神经元相关成分的肽能细胞类型,例如源自具有神经发生特征的祖细胞的突触前支架。相比之下,海绵和栉水母等早期分枝动物则缺乏这种保守的表达。
- 我们的研究结果表明,关键的神经元发育和效应基因模块在旁分泌细胞信号传导背景下的刺胞动物/两侧对称动物神经元出现之前就已经进化。
婴儿哭声诱导母体催产素释放的神经回路
Neural circuitry for maternal oxytocin release induced by infant cries. Nature
之前还以为这个现象的机制已经被解决了
- 催产素是一种神经肽,对于孕产妇生理和儿童保育(包括分娩和哺乳期间的排乳)非常重要。哺乳会触发催产素的释放,但其他感觉线索(特别是婴儿的哭声)可以增加新妈妈的催产素水平,这表明哭声可以激活下丘脑催产素神经元。
- 在这里,我们描述了一种将婴儿发声的听觉信息传递给小鼠催产素神经元的神经回路。我们对清醒母鼠中已识别的催产素神经元进行了体内电生理记录和光度测定,这些母鼠发出幼崽的叫声。我们发现,通过后层内丘脑的输入,催产素神经元对幼崽的发声做出反应,但对纯音没有反应,并且重复的丘脑刺激会诱导催产素神经元的持久去抑制。
- 该电路控制中枢催产素的释放和母体响应呼叫的行为,提供了一种将后代的感觉线索整合到母体内分泌网络中的机制,以确保大脑状态的调节,从而实现有效的育儿。
颅缝早闭和颅骨矿化的多干细胞基础
A multi-stem cell basis for craniosynostosis and calvarial mineralization. Nature
- 颅缝早闭是一组颅骨缝过早融合的疾病。在颅缝早闭中产生融合驱动成骨细胞的颅骨干细胞(CSC)的身份仍然知之甚少。
- 在这里,我们发现颅缝早闭中的生理性颅骨矿化和病理性颅骨融合反映了两个独立干细胞谱系的相互作用;我们在本研究中鉴定了先前鉴定的组织蛋白酶 K (CTSK) 谱系 CSC1 (CTSK+ CSC) 和含有盘状蛋白结构域的受体 2 (DDR2) 谱系干细胞 (DDR2+ CSC)。 Twist1(一种与人类颅缝早闭相关的基因)仅在 CTSK+ CSC 中缺失就足以驱动小鼠颅缝早闭,但注定要融合的位点表现出 CTSK+ CSC 的意外耗尽和 DDR2+ CSC 的相应扩张, DDR2+ CSC 扩展是对 CTSK+ CSC 耗尽的直接适应不良反应。 DDR2+ CSC 显示出完整的干细胞特征,
- 我们的结果证实了缝线中存在两种不同的干细胞谱系,这两种细胞群都有助于生理性颅骨矿化。 DDR2+ CSC 介导一种独特形式的软骨内骨化,而没有典型的造血骨髓形成。将 DDR2+ CSC 植入缝合位点足以诱导融合,并且通过 CTSK+ CSC 的共移植来防止这种表型。最后,DDR2+ CSC 和 CTSK+ CSC 的人类对应物在异种移植测定中显示出保守的功能特性。
- 这两种干细胞群之间的相互作用为调节颅骨矿化和缝线通畅提供了新的生物界面。
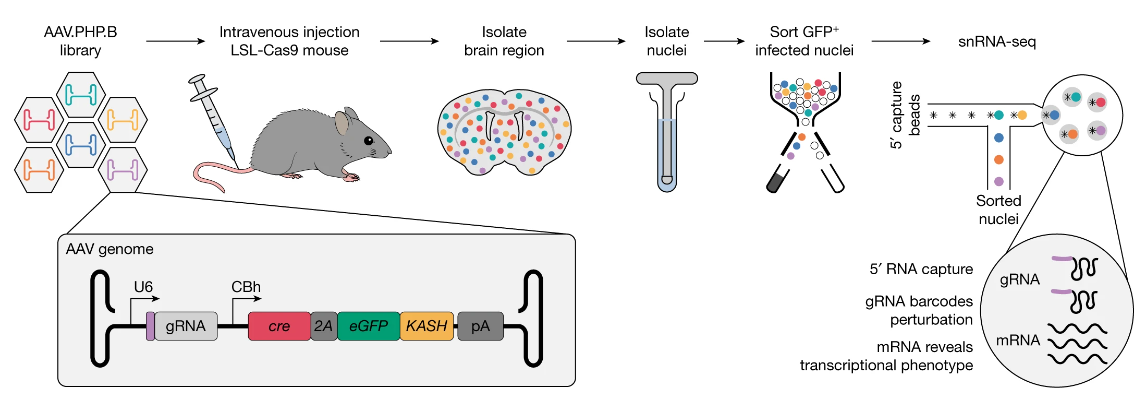
(~ ̄▽ ̄)~ 使用体内 AAV-Perturb-seq 进行转录连锁分析
Transcriptional linkage analysis with in vivo AAV-Perturb-seq. Nature
了解一下研究方法
- 与人类病理学相关的遗传变异的概要不断增长,需要新的方法以高通量的方式研究复杂组织中的基因型-表型关系。
- 在这里,我们介绍腺相关病毒(AAV)介导的直接体内单细胞 CRISPR 筛选,称为 AAV-Perturb-seq,这是一种可调节且广泛适用的方法,用于体内转录连锁分析以及高通量和高分辨率的遗传扰动表型分析。
- 我们应用 AAV-Perturb-seq 使用基因编辑和转录抑制来系统地剖析成年小鼠大脑前额皮质中 22q11.2 缺失综合征基因的表型景观。我们鉴定了三个 22q11.2 连锁基因,这些基因参与已知和以前未描述的体内协调神经元功能的途径,这解释了在 22q11.2 缺失小鼠模型中观察到的大约 40% 的转录变化。我们的研究结果表明,在成熟神经元中发现的 22q11.2 缺失综合征转录表型可能部分归因于与疾病易感性相关的一类基因的广泛失调,这些基因对于功能失调的 RNA 加工和突触功能非常重要。
- 我们的研究建立了一种灵活且可扩展的直接体内方法,以促进对生物学和疾病机制的因果理解,并具有潜在的应用来确定治疗疾病的遗传干预和治疗靶点。
β1-肾上腺素能受体将交感神经与 T 细胞耗竭联系起来
The β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature
已添加GSClassifier-PAD背景
- CD8+ T细胞是针对病毒感染和肿瘤的免疫反应的重要组成部分,并且能够消除感染细胞和癌细胞。然而,当抗原无法被清除时,T细胞就会进入一种称为衰竭的状态。尽管很明显慢性抗原会导致 CD8+ T 细胞耗竭,但人们对组织中的应激反应如何调节 T 细胞功能知之甚少。
- 在这里,我们通过 β1-肾上腺素能受体 ADRB1 展示了压力相关儿茶酚胺与 T 细胞耗竭进展之间的新联系。我们发现,耗尽的 CD8+ T 细胞会增加 ADRB1 的表达,而 ADRB1+ T 细胞暴露于儿茶酚胺会抑制其细胞因子的产生和增殖。耗尽的 CD8+ T 细胞以 ADRB1 依赖性方式聚集在交感神经周围。 β1-肾上腺素能信号传导的消除限制了慢性感染中 T 细胞向衰竭状态的进展,并在与黑色素瘤中的免疫检查点阻断 (ICB) 相结合时改善了效应器功能。
- 在对 ICB 耐药的胰腺癌模型中,β 受体阻滞剂和 ICB 协同增强 CD8+ T 细胞反应并诱导组织驻留记忆样 T 细胞的发育。恶性肿瘤与患者儿茶酚胺水平升高相关,我们的结果建立了交感应激反应、组织神经支配和 T 细胞耗竭之间的联系。
- 在这里,我们发现了一种新机制,通过阻断 CD8+ T 细胞中的 β-肾上腺素信号传导可以恢复抗肿瘤功能。
转基因雪貂模型定义了肺离子细胞的多样性和功能
Transgenic ferret models define pulmonary ionocyte diversity and function. Nature
怎么想到要用雪貂来做实验的呢 (ฅ´ω`ฅ)
- 物种形成导致器官细胞生理学的适应性变化,并为研究人类和小鼠之间存在差异的稀有细胞类型功能带来了挑战。罕见的富含囊性纤维化跨膜电导调节因子 (CFTR) 的肺离子细胞存在于人类的软骨气道中,但小鼠近端气管中的有限存在和不同的生物学特性阻碍了使用传统转基因模型来阐明气道中离子细胞的功能。
- 在这里,我们描述了条件遗传雪貂模型的创建和使用,通过启用离子细胞谱系追踪(FOXI1-CreERT2::ROSA-TG)、离子细胞消融(FOXI1-KO)和离子细胞特异性删除 CFTR 来剖析肺离子细胞生物学和功能(FOXI1-CreERT2::CFTRL/L)。
- 通过将这些模型与囊性纤维化雪貂进行比较,我们证明离子细胞控制气道表面液体吸收、分泌、pH和粘液粘度,导致囊性纤维化、FOXI1-KO和FOXI1-CreERT2中气道表面液体量减少和粘膜纤毛清除受损::CFTRL/L 雪貂。这些过程受 CFTR 依赖性离子细胞 Cl− 和 HCO3− 转运的调节。单细胞转录组学和体内谱系追踪揭示了气道发育过程中肺离子细胞的三种亚型以及离子细胞、簇细胞和神经内分泌细胞的 FOXI1 谱系常见稀有细胞祖细胞。因此,罕见的肺离子细胞在近端气道中执行关键的 CFTR 依赖性功能,这是囊性纤维化气道疾病的标志性特征。
- 这些研究为在第一种非啮齿类哺乳动物中使用条件遗传学来解决人类和雪貂之间具有更大进化保守性的基因功能、细胞生物学和疾病过程提供了路线图。
波亚胺的合成揭示了其抗癌活性的基础
Synthesis of portimines reveals the basis of their anti-cancer activity. Nature
斯克里普斯研究中心化学系
- 海洋来源的环状亚胺毒素 Portimine A 和 Portimine B 因其化学结构和显着的抗癌治疗潜力而引起人们的关注。然而,目前获取大量这些毒素是不可行的,并且其有效活性的分子机制至今仍不清楚。
- 为了解决这个问题,提出了一种可扩展且简洁的波尔亚明合成方法,该合成方法受益于两相萜类化合物合成中使用的逻辑以及其他策略,例如利用环链互变异构和骨架重组,以通过自保护最小化保护基团化学反应。
- 值得注意的是,这种全合成实现了 portimine B 的结构重新分配和对 portimine A 的深入功能评估,揭示了它在人类癌细胞系中高效选择性诱导细胞凋亡,并且在体内肿瘤清除模型中有效。
- 最后,波替明及其类似物的实际应用简化了光亲和类似物的开发,这些类似物被用于化学蛋白质组学实验,以确定波替明 A 的主要靶标为 60S 核糖体输出蛋白 NMD3。
乙酰甲基赖氨酸在活性转录起始位点标记染色质
Acetyl-methyllysine marks chromatin at active transcription start sites. Nature
- 组蛋白和其他蛋白质中的赖氨酸残基可以通过编码调控信息的翻译后修饰进行修饰。赖氨酸乙酰化和甲基化对于调节染色质和基因表达尤其重要。涉及这些翻译后修饰的途径是临床批准的治疗人类疾病的疗法的目标。通常认为赖氨酸甲基化和乙酰化在同一残基上是相互排斥的。
- 在这里,我们报告了细胞赖氨酸残基在同一侧链上被甲基化和乙酰化,形成 Nε-乙酰基-Nε-甲基赖氨酸 (Kacme)。我们发现 Kacme 存在于一系列物种和哺乳动物组织的组蛋白 H4 (H4Kacme) 上。 Kacme 与活性染色质标记、转录起始增加相关,并根据生物信号进行调节。
- H4Kacme 可以通过单甲基赖氨酸肽的酶促乙酰化来安装,并且在体外能够抵抗某些 HDAC 的脱乙酰化。 Kacme 可以与识别修饰的赖氨酸残基的染色质蛋白结合,正如我们通过与组蛋白 H4Kacme 肽结合的乙酰赖氨酸结合蛋白 BRD2 的晶体结构所证明的那样。
- 这些结果确立了 Kacme 作为一种细胞翻译后修饰,具有编码与单独甲基化和乙酰化不同的信息的潜力,并证明 Kacme 具有翻译后修饰的所有特征,对染色质生物学具有根本重要性。
(~ ̄▽ ̄)~ 抑制IDH2通过激活补偿代谢途径促进记忆 CD8+ T 细胞分化,但不影响T细胞增殖及效应功能
Reductive carboxylation epigenetically instructs T cell differentiation. Nature
洛桑大学肿瘤学系
很nice的研究发现,对CAR-T的设计具有重要启示作用
- 针对病原体或癌症的保护性免疫是通过抗原特异性初始 T 细胞的激活和克隆扩增为效应 T 细胞来介导的。为了维持其快速增殖和效应功能,初始 T 细胞通过增加有氧糖酵解水平,同时通过线粒体代谢和氧化磷酸化,将其静态代谢转变为合成代谢,产生能量和信号分子。然而,代谢重编程(metabolic rewiring)如何驱动和定义 T 细胞的分化仍不清楚。
- 在这里,我们展示了增殖效应 CD8+ T 细胞通过线粒体酶异柠檬酸脱氢酶 2 (IDH2) 还原性羧化谷氨酰胺。值得注意的是,编码 IDH2 的基因的缺失不会损害 T 细胞的增殖及其效应功能,但会促进记忆 CD8+ T 细胞的分化。因此,在嵌合抗原受体(CAR)T细胞的离体制造过程中抑制IDH2可诱导记忆T细胞的特征,并增强黑色素瘤、白血病和多发性骨髓瘤的抗肿瘤活性。
- 从机制上讲,抑制 IDH2 会激活补偿代谢途径,导致调节组蛋白修饰酶的代谢物不平衡,从而维持记忆 T 细胞分化所需基因的染色质可及性。
- 这些发现表明,CD8+ T 细胞中的还原羧化对于其效应器反应和增殖来说是可有可无的,但它主要产生一种代谢物模式,该模式在表观遗传上将 CD8+ T 细胞锁定在终末效应器分化程序中。阻断这一代谢途径可以增加记忆 T 细胞的形成,从而可以用来优化 CAR T 细胞的治疗效果。
基于AI的深部脑刺激疗效的标志物
Cingulate dynamics track depression recovery with deep brain stimulation. Nature
看上去有点神奇
- 胼胝体下扣带回 (SCC) 的深部脑刺激 (DBS) 可以长期缓解难治性抑郁症 (TRD) 的症状。然而,实现稳定恢复是不可预测的,通常需要根据个人恢复轨迹和主观症状报告进行试错刺激调整。我们目前缺乏客观的基于大脑的生物标志物来通过区分自然短暂的情绪波动和需要干预的情况来指导临床决策。
- 为了解决这一差距,我们使用了一种支持电生理学记录的新设备,为 10 名 TRD 参与者提供 SCC DBS(ClinicalTrials.gov 标识符 NCT01984710)。在 24 周的研究终点,90% 的参与者表现出强劲的临床反应,70% 的参与者获得缓解。
- 利用六位参与者提供的 SCC 局部场电位,我们部署了一种可解释的人工智能方法来识别 SCC 局部场电位变化,表明患者当前的临床状态。这种生物标志物与瞬态刺激效应不同,对治疗调整敏感,并且能够准确捕捉个体恢复状态。根据目标白质治疗网络内结构完整性和功能连接的术前损伤程度来预测可变的恢复轨迹,并与使用数据驱动的视频分析检测到的客观面部表情变化进行匹配。
- 我们的结果证明了客观生物标志物在个性化 SCC DBS 管理中的实用性,并为 TRD 病理学多方面(功能、解剖和行为)特征之间的关系提供了新的见解,推动了对抑郁症治疗变异原因的进一步研究。
Mpox 感染可防止恒河猴再次受到攻击
Mpox infection protects against re-challenge in rhesus macaques. Cell
这种表现类似于许多病毒的感染,初次感染后已建立起免疫保护屏障
- 2022 年至 2023 年爆发的 MPOX 疫情在全球男男性行为者中迅速传播。
- 我们通过静脉内、皮内和直肠内途径感染了 18 只恒河猴,并在所有三种感染途径后观察到了强烈的抗体和 T 细胞反应。静脉内和皮内感染后观察到许多皮肤损伤和高血浆病毒载量。皮肤损伤在感染后第 10 天达到顶峰,并在第 28 天消退。
- 第 28 天,我们再次对所有恢复期动物和 3 只幼稚动物进行 MPOX 攻击。所有恢复期动物均受到保护,免受再次攻击。
- 转录组学研究表明,攻击后先天反应和炎症反应上调,胶原蛋白形成和细胞外基质组织下调,以及再次攻击后 T 细胞和浆细胞反应快速激活。
- 这些数据揭示了MPOX发病机制和免疫的关键机制。这种猕猴模型应该有助于评估MPOX疫苗和治疗方法。
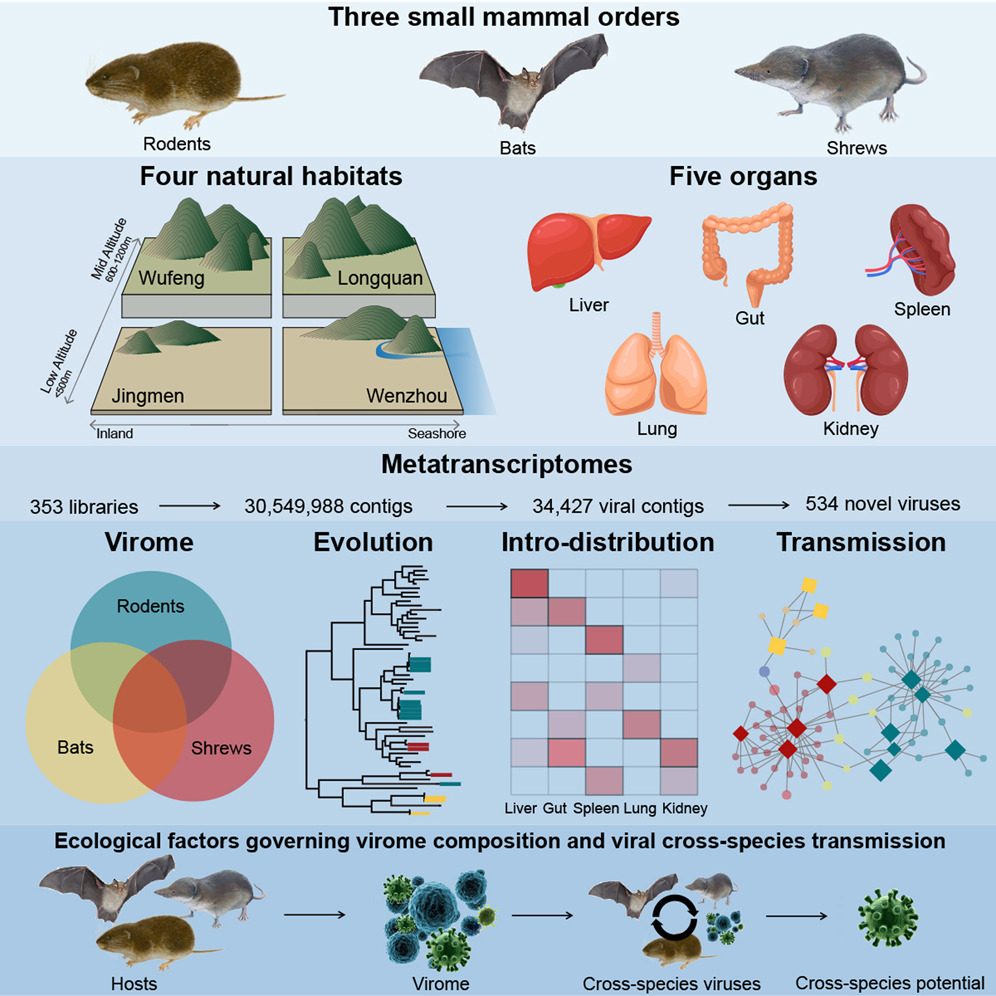
宿主特征影响野生小型哺乳动物的病毒组组成和病毒传播
Host traits shape virome composition and virus transmission in wild small mammals. Cell
复旦大学大湾区精准医学研究院(广州)基因工程国家重点实验室
- 蝙蝠、啮齿动物和鼩鼱是人类传染病最重要的动物来源。然而,病毒在它们之间的进化和传播在很大程度上仍未被探索。
- 通过对来自中国四个栖息地的2,443份野生蝙蝠、啮齿动物和鼩鼱的内脏和粪便样本进行宏转录组测序,鉴定出669种病毒,其中包括534种新病毒,从而极大地扩展了哺乳动物病毒组。
- 我们的分析揭示了高水平的系统发育多样性,确定了跨物种病毒传播事件,阐明了病毒起源,并确定了哺乳动物宿主中的无脊椎动物病毒病例。宿主顺序和样本大小是影响病毒组组成和病毒溢出模式的最重要因素。
- 鼩鼱携带着高度丰富的病毒,包括许多与多器官分布的无脊椎动物相关的病毒,而啮齿类动物携带的病毒具有更强的宿主跳跃能力。
- 这些数据突显了当地栖息地中哺乳动物病毒的显着多样性及其在新宿主中出现的能力。
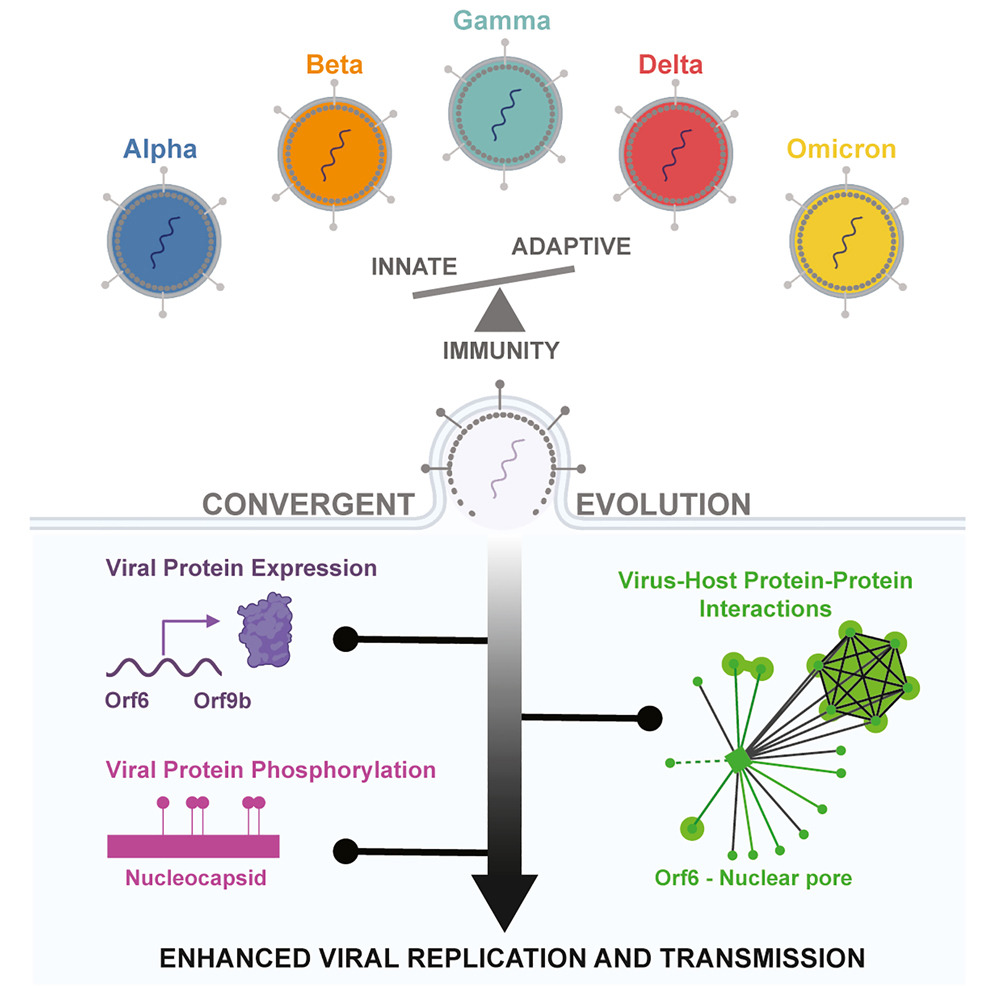
SARS-CoV-2 变体进化出收敛策略来重塑宿主反应
SARS-CoV-2 variants evolve convergent strategies to remodel the host response. Cell
- COVID-19 大流行期间出现了 SARS-CoV-2 相关变种 (VOC)。在这里,我们使用无偏系统方法来研究驱动 VOC 演化的宿主选择力。
- 我们发现,VOC 进化出了聚合策略,通过调节病毒 RNA 和蛋白质水平、改变病毒和宿主蛋白质磷酸化以及重新连接病毒-宿主蛋白质-蛋白质相互作用来重塑宿主。
- 综合计算分析表明,虽然 Alpha、Beta、Gamma 和 Delta 最终收敛到抑制干扰素刺激基因 (ISG),但 Omicron BA.1 却没有。 ISG 抑制与病毒先天免疫拮抗蛋白的表达相关,包括 Orf6、N 和 Orf9b,我们将其映射到特定突变。后来的 Omicron 亚变体 BA.4 和 BA.5 比早期亚变体 BA.1 更有效地抑制先天免疫,后者与 Orf6 水平相关,尽管 BA.4 中的突变因破坏 Orf6-核孔相互作用而减弱。
- 我们的研究结果表明,SARS-CoV-2趋同进化克服了人类适应性和先天免疫障碍,为应对未来的流行病奠定了基础。
通过将特征神经元再生到其自然目标区域来恢复瘫痪后行走
似乎是一个很自然的假设,没想到在最近才得到验证
- 轴突再生可以在解剖上完全脊髓损伤(SCI)中诱导,但稳健的功能恢复一直难以实现。恢复神经功能是否需要轴突从特定神经元亚群定向再生到其自然目标区域仍不清楚。
- 为了解决这个问题,我们应用投影特异性和比较单核 RNA 测序来识别在不完全 SCI 后恢复行走的神经元亚群。我们发现,化学吸引和引导这些神经元横断的轴突到达其自然目标区域,导致小鼠在完全脊髓损伤后行走能力得到显着恢复,而轴突仅穿过病变部位的再生则没有效果。
- 因此,重建特征神经元的自然投射构成了旨在恢复丢失的神经功能的轴突再生策略的重要组成部分。
妊娠诱导先前存在的微嵌合细胞移位后的生殖结果
Reproductive outcomes after pregnancy-induced displacement of preexisting microchimeric cells. Science
- 怀孕可以为伴侣提供针对未来怀孕并发症的保护,这与分娩后母亲体内胎儿微嵌合细胞(FMcs)的持续存在类似。
- 我们发现,怀孕期间先前存在的 FMc 会被新的 FMc 取代,并且 FMc 强直刺激对于保护性胎儿特异性叉头盒 P3 (FOXP3) 阳性调节 T 细胞 (Treg 细胞) 的扩增至关重要。
- 怀孕后,母体微嵌合细胞和具有非遗传性母体抗原 (NIMA) 特异性的 Treg 细胞的积累在女儿身上也发生了类似的逆转,突出了固定的微嵌合细胞生态位。虽然 NIMA 特异性耐受性会因怀孕而在功能上消失,但尽管怀孕期间亲子关系发生了变化,但母亲对妊娠并发症的针对伴侣特异性的弹性仍然存在。
- 持久的胎儿耐受性反映了 FOXP3 表达的可塑性,这使得母亲能够更持久地记住她们的婴儿,而女儿则通过新的妊娠印记免疫记忆忘记她们的母亲。
操纵线粒体电子流增强肿瘤免疫原性
Manipulating mitochondrial electron flow enhances tumor immunogenicity. Science
- 尽管肿瘤生长需要线粒体电子传递链 (ETC),但复合物 I (CI) 和复合物 II (CII)(启动电子流的看门人)的相对贡献仍不清楚。
- 在这项工作中,我们报告说,CII 的缺失(而非 CI 的缺失)通过增加抗原呈递和 T 细胞介导的杀伤来减少黑色素瘤的生长。这是由琥珀酸介导的主要组织相容性复合体-抗原加工和呈递(MHC-APP)基因的转录和表观遗传激活驱动的,与干扰素信号传导无关。
- 此外,敲除甲基化控制的 J 蛋白 (MCJ),以促进电子优先通过 CI 进入,提供了 ETC 重新布线的概念证明,以实现抗肿瘤反应,且不会产生与非癌细胞线粒体呼吸总体减少相关的副作用。
- 我们的结果可能对 MHC-APP 表达减少的肿瘤具有治疗潜力,这是癌症免疫逃避的常见机制。
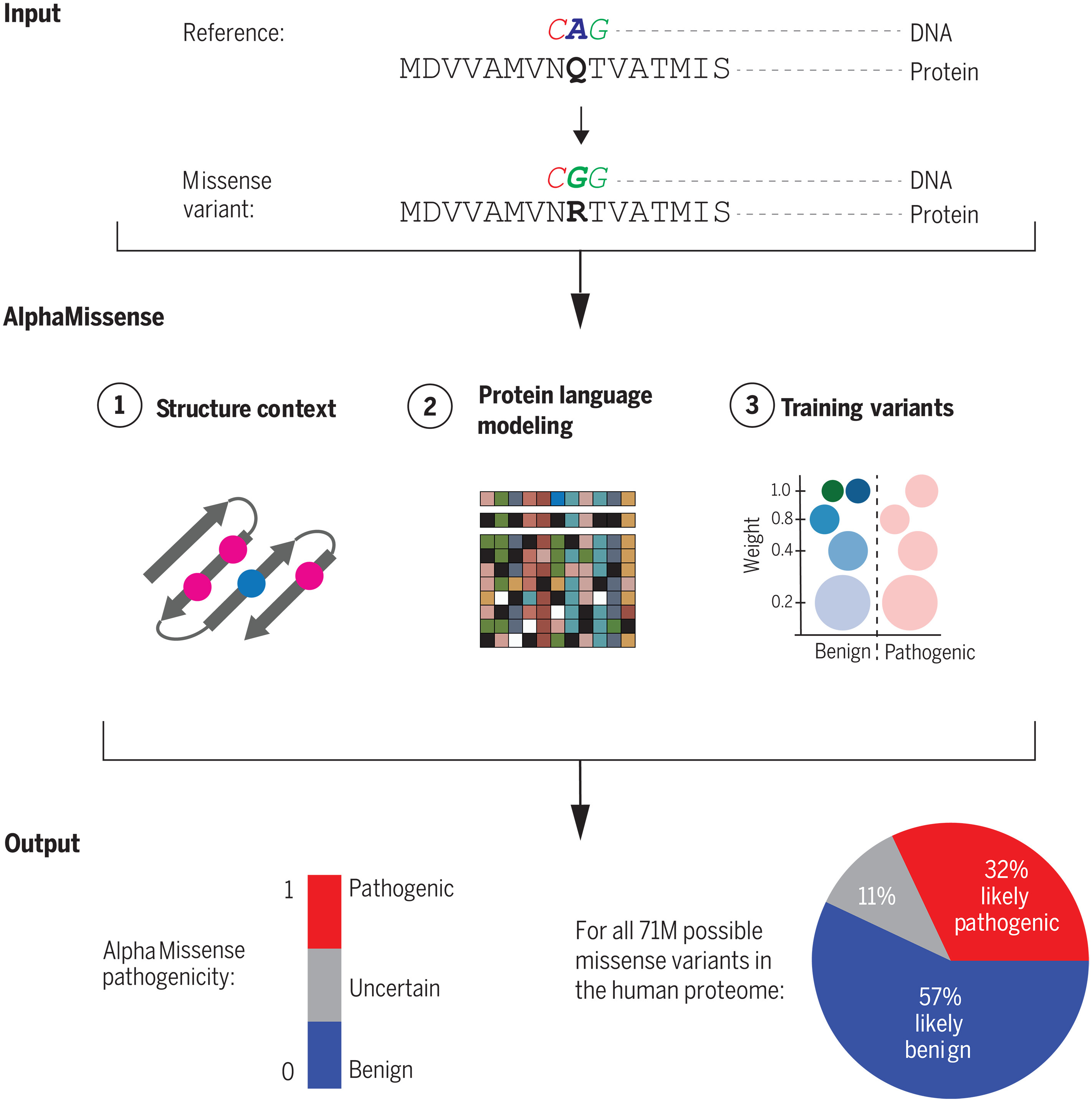
(~ ̄▽ ̄)~ 使用 AlphaMissense 准确预测整个蛋白质组错义变异效应
Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. full html
DeepMind团队
了解一下AlphaMissense的具体工作细节
- 基因组测序揭示了人类群体中广泛的遗传变异。错义变异是改变蛋白质氨基酸序列的遗传变异。致病性错义变异会破坏蛋白质功能并降低机体适应性,而良性错义变异的作用有限。
- 对这些变异进行分类是人类遗传学中一个重要的持续挑战。在观察到的超过 400 万个错义变异中,估计只有 2% 被临床分类为致病性或良性,而其中绝大多数具有未知的临床意义。这限制了罕见疾病的诊断,以及针对潜在遗传原因的临床治疗的开发或应用。机器学习方法可以通过利用生物数据中的模式来预测未注释变异的致病性,从而缩小变异解释差距。具体来说,AlphaFold可以根据蛋白质序列准确预测蛋白质结构,可以作为预测蛋白质变异体致病性的基础。
- 我们开发 AlphaMissense 是为了利用多个方面的进步:(i)无监督蛋白质语言模型,用于学习以序列上下文为条件的氨基酸分布; (ii) 通过使用 AlphaFold 衍生系统纳入结构背景; (iii) 对人口频率数据中的弱标签进行微调,从而避免人工注释带来的偏差。
- AlphaMissense 在临床注释、新发疾病变异和实验检测基准方面实现了最先进的错义致病性预测,而无需对此类数据进行明确的训练。作为社区的资源,我们提供了人类蛋白质组中所有可能的单一氨基酸取代的预测数据库。我们使用 ClinVar 数据集上的精确度为 90% 的截止值将所有错义变异中的 32% 分类为可能致病,将 57% 分类为可能良性,从而为大多数人类错义变异提供可靠的预测。
- 我们展示了如何利用该资源来加速多个领域的研究。分子生物学家可以使用该数据库作为设计和解释实验的起点,以探测人类蛋白质组中的饱和氨基酸取代。人类遗传学家可以将基因水平的 AlphaMissense 预测与基于群体队列的方法结合起来,以量化基因的功能意义,特别是对于较短的人类基因,因为基于队列的方法缺乏统计能力。最后,在优先考虑罕见疾病诊断的从头变异时,临床医生可以受益于可靠分类的致病变异覆盖范围的增加,而 AlphaMissense 预测可以为使用罕见、可能有害变异注释的复杂性状遗传学研究提供信息。
- AlphaMissense 预测可以阐明变异对蛋白质功能的分子影响,有助于识别致病性错义突变和以前未知的致病基因,并提高罕见遗传病的诊断率。 AlphaMissense 还将促进从结构预测模型进一步开发专门的蛋白质变异效应预测器。
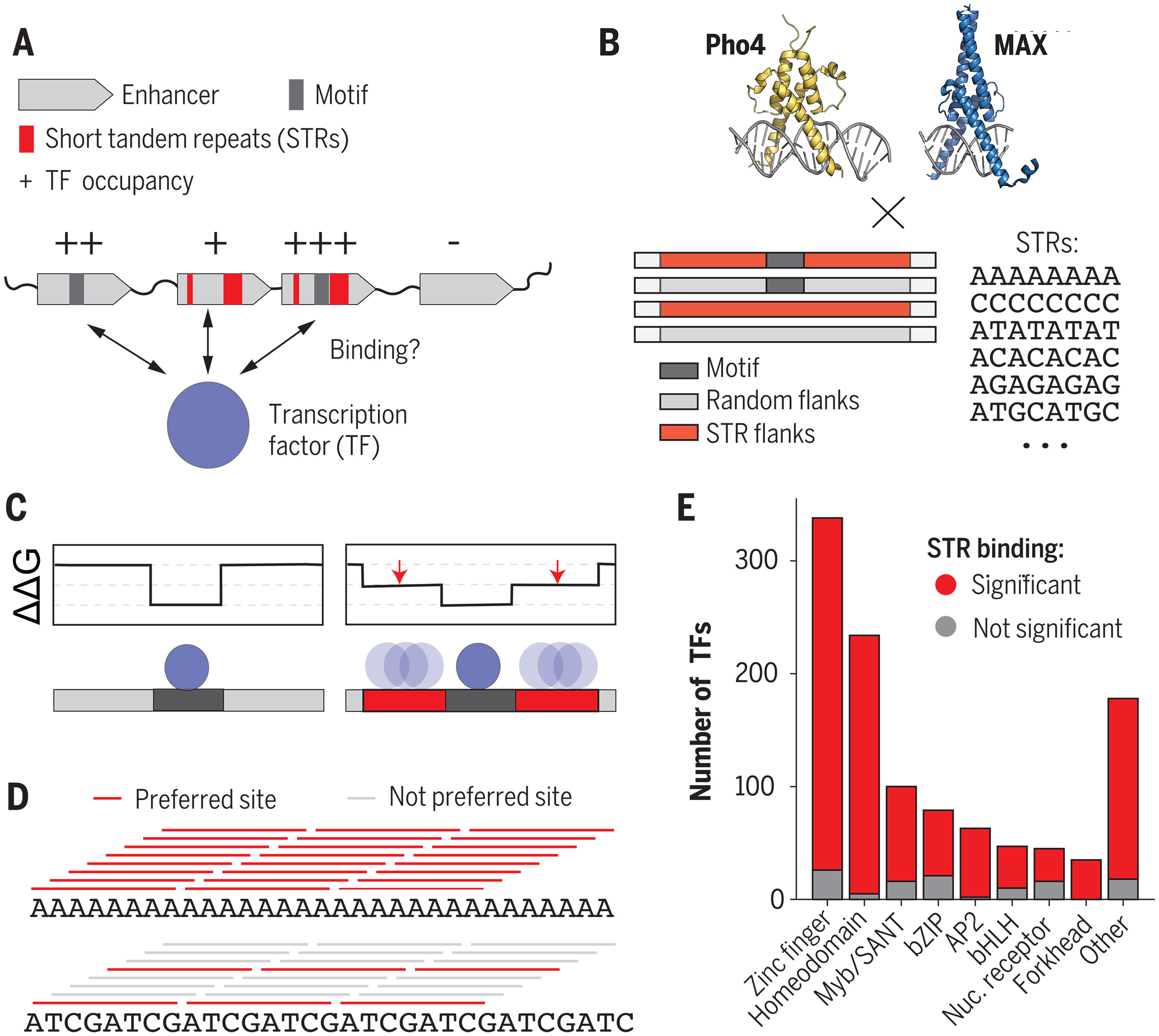
短串联重复序列结合转录因子来调节真核基因表达
Short tandem repeats bind transcription factors to tune eukaryotic gene expression. Science
- 基因表达受转录因子 (TF) 蛋白的调节,这些蛋白与基因组中的 DNA 调节元件结合。尽管数十年的研究对 TF“基序”进行了分类,但这些并不能完全解释在细胞中观察到的基因组结合。许多 TF 结合缺乏基序的区域,而其他具有明显强基序的区域仍然未被占据,并且新出现的证据表明基序周围的 DNA 序列背景可以强烈影响结合(参见图 A)。短串联重复序列(STR,一到六个核苷酸的连续重复单元)提供了这些序列背景的一个很好的例子。 STR 约占人类基因组的 5%(相比之下,所有蛋白质编码基因仅占 1.5%),并且富含增强子。 STR 长度的变化与基因表达的变化有关,并与几种复杂的表型有关,例如精神分裂症、癌症、自闭症和克罗恩病。然而,STR 影响转录的机制仍不清楚。
- STR 影响基因表达的一种机制是改变 TF 与调节 DNA 结合的亲和力和/或动力学(见图 A 部分)。为了研究这一点,我们使用了各种高通量微流体结合测定(即 MITOMI、k-MITOMI 和 STAMMP)和生物信息学分析来系统地量化不同序列背景对 TF 结合的影响。我们测量了两个基本螺旋-环-螺旋 TF 的亲和力 (Kds) 和动力学 (koffs),它们将 CACGTG E-box 基序(来自酿酒酵母的 Pho4 和来自智人的 MAX)与带有或不带有 E-box 基序的 DNA 序列结合被随机序列或多种不同类型的 STR 包围(见图 B 组)。
- 测量的 609 个不同 TF-DNA 组合的结合常数 (Kds) 表明,不同的 STR 可以将结合亲和力改变 > 70 倍(见图,图 C),接近或超过突变共有基序的效果。 Pho4 和 MAX TF 的首选 STR 不同,表明基序不足以预测首选 STR。凝胶迁移测定和使用 TF 截短构建体的其他实验证实,在存在或不存在基序的情况下,TF 通过其 DNA 结合域直接结合 STR(见图,C 组)。尽管没有通过标准单核苷酸模型进行预测,但观察到的 STR 结合可以通过统计力学的简单分配函数模型得到很好的解释,其中多个重复的弱结合位点对结合亲和力有附加作用(见图 D)。测量的 106 个 TF-DNA 组合的表观解离率 (koffs) 和动力学模型表明,STR 主要改变宏观表观关联率并增加 DNA 结合 TF 的局部密度。最后,仅根据体内全基因组染色质免疫沉淀数据训练的神经网络预测的效果与体外测量的效果相同,这表明 STR 偏好在细胞中正确定位 TF 方面发挥着重要作用。
- 总之,对先前发表的蛋白质结合微阵列和 SELEX 数据的分析表明,约 90% 的真核 TF 优先结合至少一种类型的 STR(见图 E 组)。由于 STR 具有高度可变性,我们建议将它们视为一类易于进化的顺式调控元件。优选的 STR 不需要与已知基序相似,这表明可以将 TF 旁系同源物募集到不同的调控区域并调控不同的靶基因。尽管 STR 最大限度地增加了潜在弱结合位点的数量,但我们预计包含许多低亲和力结合位点的非重复序列上下文应该类似地增加结合。因此,我们提出 STR 作为“变阻器”来调节局部 TF 浓度和结合反应,从而调节疾病、发育和稳态中的基因表达。
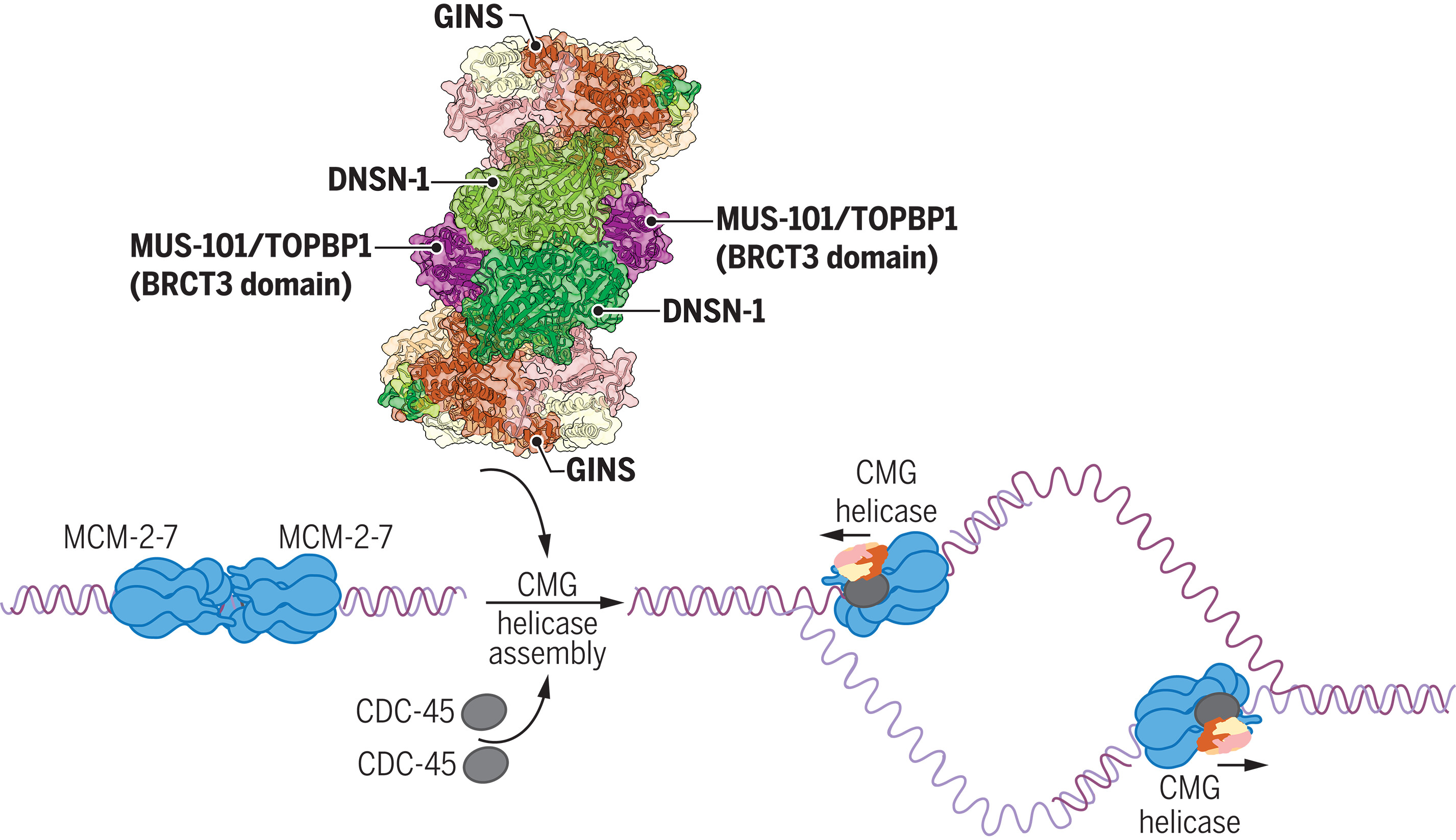
DNSN-1 在秀丽隐杆线虫 DNA 复制启动过程中招募 GINS 进行 CMG 解旋酶组装
系列研究
- 真核染色体由称为复制体的分子机器复制,复制体的组装受到高度调控,以确保细胞在每个细胞周期中制作其基因组的单个副本。在人类中,复制体组装的缺陷通常与早期癌症的发展有关,并可能导致一种称为迈耶-戈林综合征的小头型原始侏儒症。真核复制体组装是由称为 CMG (CDC-45–MCM-2-7–GINS) 的 11 亚基解旋酶的组装和激活启动的,复制体围绕该解旋酶形成。首先,称为 MCM-2-7(包含 MCM-2 至 MCM-7 蛋白)的六种腺苷三磷酸酶 (ATPase) 的两个环在双链 DNA 周围组装,在有丝分裂结束时在复制起点形成双六聚体。其次,CDC-45 和四蛋白 GINS 复合物在 S 期被招募至 MCM-2-7 双六聚体,在两个新生复制体的中心形成一对 CMG 解旋酶,这一过程由多种蛋白激酶控制和多个组装因素。最后,两个解旋酶在一个鲜为人知的步骤中被激活,其中 MCM-2-7 环被瞬时打开以排除一条 DNA 链。
- CMG 解旋酶组装和激活的机制已使用芽殖酵母进行了最深入的研究,其中 DNA 复制的整个周期已用纯化的蛋白质重建。然而,最近的证据表明介导和控制 CMG 组装的因素存在相当大的进化多样性。例如,Cdc7 激酶对于芽殖酵母中解旋酶的组装至关重要,但 CDC7 在未转化的小鼠和人类细胞中却是可有可无的。此外,酵母Sld2蛋白是一种重要的解旋酶组装因子,与脊椎动物RECQL4同源,但对爪蟾RECQL4的研究表明它在CMG组装后起作用。这些发现表明动物细胞含有其他有待鉴定的解旋酶组装因子。
- 使用线虫秀丽隐杆线虫的胚胎作为研究后生动物复制体组装的模型,我们发现一种名为 DNSN-1(人类 DONSON 的直系同源物)的蛋白质对于 DNA 复制的启动至关重要,但对于复制叉的后续进展却是可有可无的。我们的数据表明,BRCT 重复蛋白 MUS-101/TOPBP1 将 DNSN-1 招募到 S 期 MCM-2-7 双六聚体上形成的预起始复合物,从而触发 CMG 解旋酶的组装。
- 我们表明,DNSN-1 对于将 CDC-45 招募到染色质来说是可有可无的,但对于 GINS 的加载却是必需的。冷冻电镜显示 DNSN-1 的二聚体同时结合到 GINS 和 MCM-3 解旋酶亚基上的多个位点,表明 DNSN-1 在 DNA 复制起始过程中定位 GINS 以促进 CMG 组装。与这一观点一致,我们表明,删除 DNSN-1 上 GINS 的一个结合位点会导致起始缺陷,而破坏与 MCM-3 的一个界面的 DNSN-1 中的点突变是致命的,并阻止 DNSN 的招募-1 为 S 期期间的预引发复合物。
在芽殖酵母中,Mcm10 蛋白是激活新生 CMG 解旋酶复合物所必需的。我们表明,通过 CRISPR-Cas9 几乎完全删除 mcm-10 编码序列在秀丽隐杆线虫中是可行的,这表明其他因素也可能有助于后生动物中解旋酶的激活。我们的数据表明 DNSN-1 也可能在此步骤中发挥作用,因为 DNSN-1 的 RNAi 消耗对 mcm-10Δ 是合成致死的。 - DNSN-1 的直系同源物存在于动物和植物中,但在芽殖酵母和许多真菌物种中不存在。这表明 DNSN-1/DONSON 在 DNA 复制起始过程中的作用在真核进化的早期阶段就出现了,但随后在真菌进化过程中消失了。
- 我们的研究结果表明,DNSN-1/DONSON 是我们理解动物细胞 DNA 复制起始过程中缺失的一环,在 CMG 解旋酶组装中发挥着重要作用。 DNA 复制起始过程中对 DNSN-1 的需求表明后生动物和芽殖酵母之间的复制体组装机制存在显着差异。与 DNSN-1 在秀丽隐杆线虫 CMG 解旋酶组装过程中的重要作用一致,人类 DONSON 突变会导致 Meier-Gorlin 综合征。
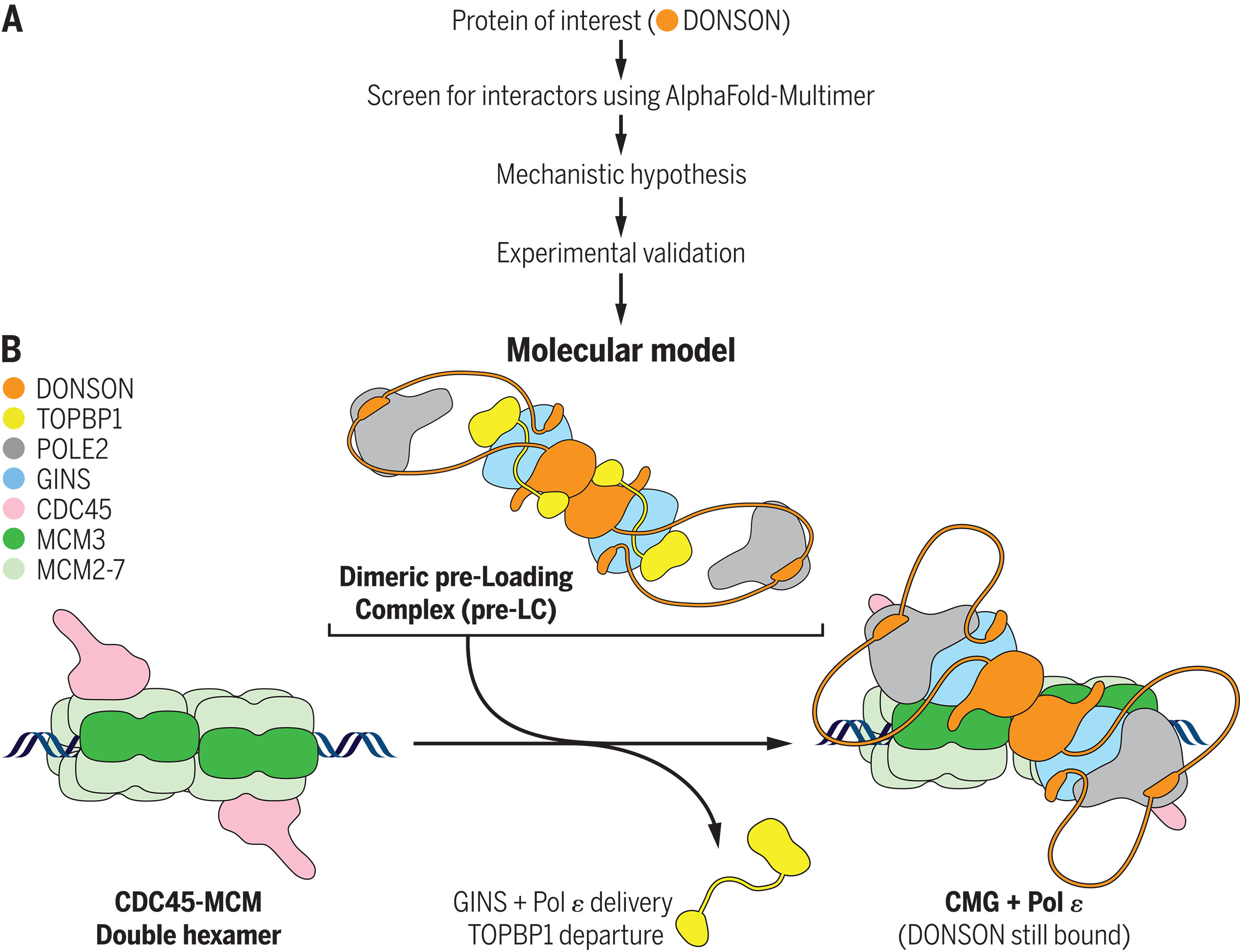
计算机模拟蛋白质相互作用筛选揭示了 DONSON 在DNA复制启动中的作用
In silico protein interaction screening uncovers DONSON’s role in replication initiation. Science
系列研究
- 快速而忠实的 DNA 复制维持了基因组的完整性,但在癌症等人类疾病中经常被破坏。复制起始过程中的一个关键事件是 CDC45-MCM2-7-GINS (CMG) 解旋酶的组装,该解旋酶在分叉处解旋 DNA,由六聚体 MCM2-7 ATP 酶和两个辅助因子 CDC45 和 GINS 组成。 MCM2-7 双六聚体被募集到 G1 起始点且细胞进入 S 期后,CDC45 和 GINS 依次与 MCM2-7 双六聚体结合,导致 CMG 组装。在酵母中,GINS 被包含 Dpb11、Sld2 和 DNA 聚合酶 ε (Pol ε) 的“预加载复合物”(pre-LC) 护送至 MCM 双六聚体,但 GINS 如何被招募到后生动物的起源仍不清楚。此外,多细胞生物体含有 SON 下游邻居 (DONSON) 等蛋白质,它们通过未知机制影响复制。
- 我们想了解在小头侏儒症中发生突变的 DONSON 如何促进 DNA 复制。为此,我们结合了生物化学、细胞生物学、小鼠遗传学和计算机筛选新的蛋白质-蛋白质相互作用。
- 我们首先从无细胞非洲爪蟾卵提取物中去除了 DONSON,它概括了基因组维护过程。我们发现,在没有 DONSON 的情况下,DNA 复制和 CMG 组装被废除。使用可诱导的 DONSON 降解决定子等位基因,我们发现 DONSON 对于人类细胞中的 CMG 组装和 DNA 复制也至关重要。
- 为了探索 DONSON 如何促进 CMG 形成,我们使用 AlphaFold-Multimer (AF-M) 在整个复制体中搜索 DONSON 相互作用蛋白。该重点通过计算机筛选确定了 GINS、TOPBP1(Dpb11 直系同源物)、POLE2(Pol ε 亚基)、MCM3(MCM2-7 亚基)和 DONSON 本身为高置信度 DONSON 相互作用因子。基于这些预测,我们假设 DONSON 支架了一个二聚体脊椎动物 pre-LC,其中包含两个 DONSON、TOPBP1、GINS 和 Pol ε 拷贝,并且该 pre-LC 将 GINS 定位在 CDC45-MCM2-7 上以进行 CMG 组装(见图)。
- 与该模型一致,生化研究证实 DONSON 与 TOPBP1 和 GINS 的相互作用对于 CMG 形成和 DNA 复制至关重要。另一方面,DONSON 与 MCM3 的相互作用对于预 LC 组装来说是可有可无的,而是促进了预 LC 对接至 MCM 双六聚体。导致人类小头侏儒症的 DONSON 突变在小鼠身上重现了这种情况,并损害了复制启动和 CMG 组装。最后,当我们使用 AF-M 和其他标准筛选整个人类蛋白质组中的 DONSON 相互作用子时,CMG 组装因子高度富集,这表明从头开始进行蛋白质功能鉴定的计算机筛选的潜力。
- 我们将 DONSON 确定为脊椎动物 CMG 组装所需的因子。它通过组织预 LC 发挥作用,将 GINS 传递到 CDC45-MCM2-7 上的结合位点,从而导致 CMG 组装。我们的结果表明,尽管所有真核生物都含有前 LC,但它们的结构不同,脊椎动物使用 DONSON 而不是 Sld2。我们的结果还表明,CMG 组装缺陷与小头侏儒症的病理生理学有关。我们认为,复制起始受损会导致细胞周期进展缓慢,从而导致这种侏儒症中出现的细胞减少。我们的结果表明,使用 AF-M 进行大规模计算机蛋白质-蛋白质相互作用筛选是识别功能相关相互作用和制定可测试的分子假设的有效方法。
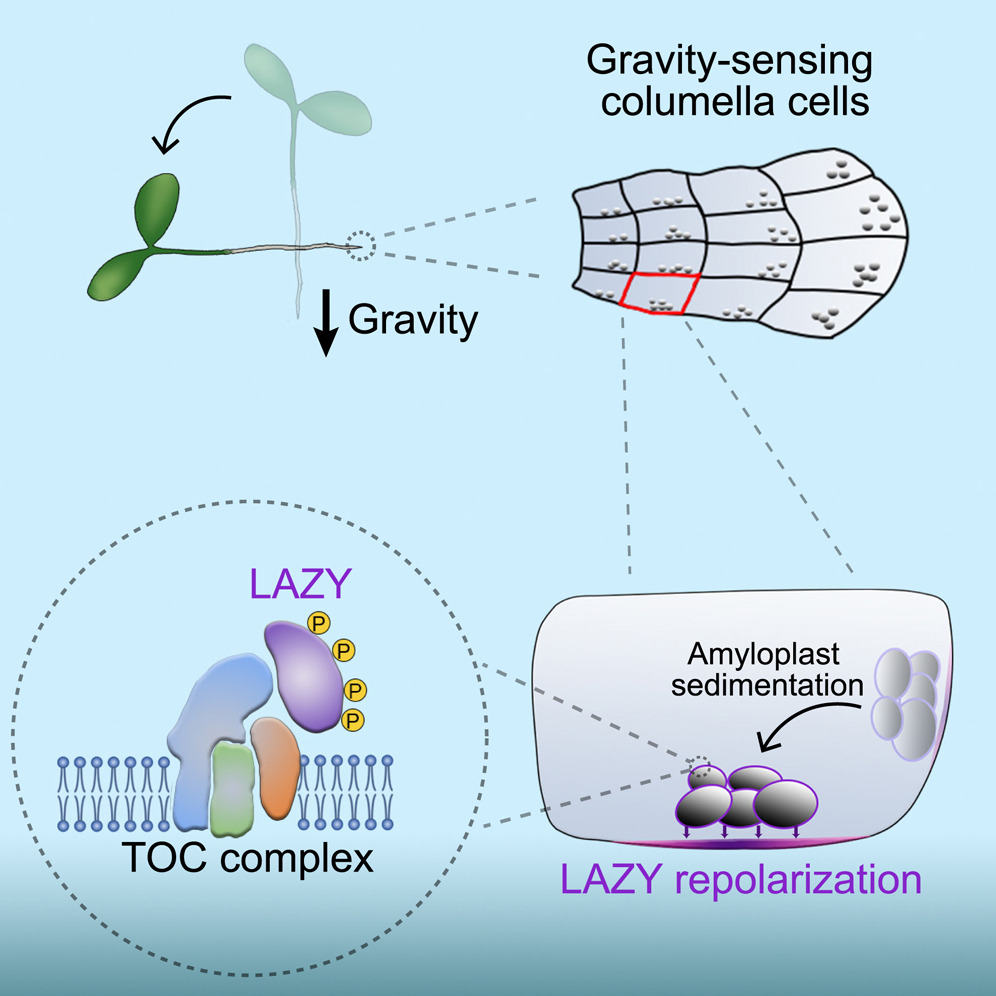
淀粉体沉降使 LAZY 重新极化以实现植物的重力感应
Amyloplast sedimentation repolarizes LAZYs to achieve gravity sensing in plants. Cell
挺有意思的研究
- 重力控制植物的定向生长,一个多世纪前提出的经典淀粉平衡石假说假设,特殊细胞中的淀粉体沉积启动了重力感应,但其分子机制仍然未知。 LAZY 蛋白被认为是向地性的关键调节因子,而惰性突变体表现出显着的向地性缺陷。
- 在这里,我们报告通过重新定向的重力刺激触发丝裂原激活蛋白激酶(MAPK)信号介导的拟南芥LAZY蛋白在根小柱细胞中基底极化的磷酸化。 LAZY 的磷酸化增加了其与淀粉体表面叶绿体 (TOC) 蛋白外膜上的几个易位子的相互作用,从而促进 LAZY 蛋白在淀粉体上的富集。淀粉体沉积随后引导 LAZY 重新定位到小柱细胞质膜的新下侧,其中 LAZY 诱导不对称的生长素分布和根部差异生长。
- 总之,这项研究为淀粉平衡石假说提供了分子解释:细胞器运动触发分子极性形成。
通过免疫分析确定“长新冠”的显着特征
Distinguishing features of Long COVID identified through immune profiling. Nature
作者如何控制混杂因素?
- 急性病毒性疾病后可能会出现急性感染后综合征 (PAIS)。感染 SARS-CoV-2 可导致 PAIS 的发展,称为“长 COVID”(LC)。 LC 患者经常报告持续疲劳、劳累后不适以及各种认知和自主神经功能障碍;然而,与这些症状的发生和持续相关的生物过程尚不清楚。
- 在此,273 名患有或未患有 LC 的个体参加了一项横断面研究,其中包括多维免疫表型分析和公正的机器学习方法,以识别与 LC 相关的生物学特征。与匹配对照相比,循环骨髓细胞和淋巴细胞群存在显着差异,并且有证据表明 LC 参与者针对 SARS-CoV-2 的体液反应过度。
- 此外,在 LC 患者中观察到针对非 SARS-CoV-2 病毒病原体的较高抗体反应,特别是 Epstein-Barr 病毒。可溶性免疫介质和激素的水平因群体而异,LC 参与者的皮质醇水平较低。将免疫表型数据整合到无偏见的机器学习模型中,确定了与 LC 状态最密切相关的关键特征。
- 总的来说,这些发现可能有助于指导未来对 LC 病理学的研究,并有助于开发相关的生物标志物。
全球 SARS-CoV-2 基因组中与 molnupiravir 相关的突变特征
A molnupiravir-associated mutational signature in global SARS-CoV-2 genomes. Nature
耐药性在病毒学研究中也不是什么新鲜事
- Molnupiravir 是一种广泛用于对抗 SARS-CoV-2 的抗病毒药物,通过在复制过程中诱导病毒基因组突变发挥作用。大多数随机突变可能对病毒有害,其中许多突变是致命的,因此莫努匹拉韦引起的突变率升高会降低病毒载量。然而,如果一些接受 molnupiravir 治疗的患者未能完全清除 SARS-CoV-2 感染,则可能存在 molnupiravir 突变病毒继续传播的可能性。
- 在这里,我们展示了 SARS-CoV-2 测序数据库包含 molnupiravir 诱变的广泛证据。使用系统方法,我们发现一类特定的长系统发育分支,以高比例的 G-to-A 和 C-to-T 突变为特征,几乎完全出现在 2022 年引入 molnupiravir 治疗后的序列中,以及广泛使用该药物的国家和年龄组。
- 我们从已知接受过莫努匹拉韦治疗的患者体内的病毒中鉴定出具有优选核苷酸背景的突变谱,并表明其特征与这些长分支中所见的特征相匹配,在某些情况下与莫努匹拉韦衍生谱系的向前传播相匹配。
- 最后,我们分析治疗记录,以确认这些高 G-to-A 分支与莫努匹拉韦的使用之间的直接关联。
自合性对整个表型谱中常见疾病风险的影响
Influence of autozygosity on common disease risk across the phenotypic spectrum. Cell
- 自合性与罕见的孟德尔疾病和临床相关的数量性状有关。我们在 Genes & Health(n = 23,978 英国南亚人)、UK Biobank(n = 397,184)和 23andMe 中研究了基因组纯合性运行比例 (the fraction of the genome in runs of homozygosity, FROH) 与常见疾病之间的关联。
- 我们表明,将分析限制于第一代表兄弟姐妹的后代是减少由于 FROH 的社会/环境相关性而造成的混杂的有效方法。在 G&H+UK Biobank 的这一组中,我们发现 FROH 与 12 种常见疾病之间存在整个实验范围内的显着关联。我们通过 23andMe 中的兄弟姐妹内分析复制了与 2 型糖尿病 (T2D) 和创伤后应激障碍的关联(中位数 n = 480,282)。我们估计,英籍巴基斯坦人中 5%–18% 的 T2D 病例是由近亲血亲导致的自合子造成的。
- 我们的工作强调了常见疾病广泛的非加性遗传效应的可能性,并对全球近亲率高的人群具有重要意义。
- Bensz:纯合性 (Runs of homozygosity, ROH) 区域是二倍体染色体的区域,其中血统相同 ( identical-by-descent, IBD) 单倍型从每个亲本遗传而来。传统上,ROH 被认为仅与近交种群相关,并且 ROH 可能与血缘关系和种群隔离有关2。
Schlafen 结构域核酸酶 PUCH 加工 piRNA
piRNA processing by a trimeric Schlafen-domain nuclease. Nature
- 转座元件是在基因组内扩展并在基因组之间传播的基因组寄生虫。 PIWI 蛋白控制转座子活性,特别是在种系中。这些蛋白质通过称为 PIWI 相互作用 RNA (piRNA) 的小 RNA 辅助因子识别其靶标,使 piRNA 生物发生成为这一关键基因组免疫系统中关键的特异性决定步骤。尽管 piRNA 前体的加工是该过程中的重要步骤,但许多分子细节仍不清楚。
- 在这里,我们鉴定了一种内切核糖核酸酶,它是 21U RNA 5′ 端裂解全酶 (PUCH) 的前体,可启动线虫秀丽隐杆线虫中的 piRNA 加工。
- 遗传和生化研究表明,PUCH 是 Schlafen 样结构域蛋白(SLFL 蛋白)的三聚体,可执行 5′ 端 piRNA 前体裂解。 PUCH 介导的加工严格需要 7-甲基-G 帽 (m7G-cap) 和第三位尿嘧啶。我们还展示了 PUCH 如何与 PETISCO(一种与 piRNA 前体结合的复合物)相互作用,并且这种相互作用增强了体内 piRNA 的产生。 PUCH 的鉴定结束了对线虫中 5′ 端 piRNA 生物合成因子的探索,并揭示了一种由三种 SLFL 蛋白形成的 RNA 核酸内切酶。
- 哺乳动物 Schlafen (SLFN) 基因与免疫相关,揭示了哺乳动物免疫反应与控制转座元件的基于深度保守的 RNA 机制之间的分子联系。
分子分辨率的小鼠中枢神经系统空间图谱
Spatial atlas of the mouse central nervous system at molecular resolution. Nature
- 在 3D 体积中以单细胞分辨率空间绘制分子细胞类型图对于说明大脑解剖结构和功能的分子基础至关重要。单细胞 RNA 测序已经分析了小鼠大脑中的分子细胞类型,但无法捕获它们的空间组织。
- 在这里,我们使用原位测序方法 STARmap PLUS 以 194 × 194 × 345 nm3 的体素大小对 1,022 个基因进行 3D 分析,绘制了成年小鼠大脑和脊髓中 109 万个高质量细胞的图谱。
- 我们开发了计算管道,通过单细胞基因表达对 230 种分子细胞类型进行分割、聚类和注释,通过空间利基基因表达对 106 个分子组织区域进行分割、聚类和注释。
- 分子细胞类型和分子组织区域的联合分析实现了系统的分子空间细胞类型命名法和对已建立的大脑解剖学中未定义的组织结构的识别。为了创建转录组范围的空间图谱,我们将 STARmap PLUS 测量结果与已发表的单细胞 RNA 测序图集1 相集成,估算 11,844 个基因的单细胞表达谱。最后,我们描述了全脑转基因递送工具 AAV-PHP.eB5,6 的病毒向性。
- 总之,这个带注释的数据集提供了一个单细胞资源,集成了分子空间图谱、大脑解剖学和哺乳动物中枢神经系统遗传操作的可访问性。
装配体 CRISPR 筛选揭示疾病基因对人类神经发育的影响
Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature
Sergiu P. Pașca团队
- 皮质回路的组装涉及中间神经元从腹侧前脑到背侧前脑的生成和迁移,这在妊娠晚期和产后早期人类发育的难以接近的阶段进行研究一直具有挑战性。自闭症谱系障碍和其他神经发育障碍 (NDD) 与皮质中间神经元发育异常有关,但哪些 NDD 基因影响中间神经元的生成和迁移,以及它们如何介导这些影响仍然未知。
- 我们之前开发了一个平台来研究大脑皮层下类器官和前脑组合体中神经元的发育和迁移。在这里,我们将组装体与 CRISPR 筛选相结合,以研究 425 个 NDD 基因在人类中间神经元发育中的参与情况。针对中间神经元生成的第一次筛选揭示了 13 个候选基因,包括 CSDE1 和 SMAD4。随后,我们对 1000 多个前脑组合体进行了中间神经元迁移筛选,确定了 33 个候选基因,包括细胞骨架相关基因和内质网相关基因 LNPK。
- 我们发现,在中间神经元迁移过程中,内质网在核易位之前沿着前导神经元分支移位。 LNPK 缺失会干扰这种内质网移位并导致异常迁移。
- 这些结果凸显了该 CRISPR 组装体平台系统地将 NDD 基因映射到人类发育并揭示疾病机制的能力。
抑制脂肪酸氧化可使成年小鼠心脏再生
Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature
“心肌细胞的不太成熟状态”与被证伪的“心肌干细胞”有哪些本质区别?
- 心肌细胞出生后成熟的特点是从糖酵解到脂肪酸氧化的代谢转变、染色质重构和退出细胞周期,为成体心脏再生设置障碍。
- 在这里,为了探索代谢重编程是否可以克服这一障碍并实现心脏再生,我们通过灭活 Cpt1b 来消除心肌细胞中的脂肪酸氧化。
- 我们发现心肌细胞脂肪酸氧化的丧失可提高对缺氧的抵抗力并刺激心肌细胞增殖,从而使缺血再灌注损伤后的心脏再生。代谢研究揭示了 Cpt1b 突变心肌细胞中能量代谢和 α-酮戊二酸积累的深刻变化,导致 α-酮戊二酸依赖性赖氨酸脱甲基酶 KDM5 的激活。
- 激活的 KDM5 会使驱动心肌细胞成熟的基因中广泛的 H3K4me3 结构域去甲基化,降低其转录水平并将心肌细胞转变为不太成熟的状态,从而促进增殖。
- 我们得出的结论是,代谢成熟塑造了心肌细胞的表观遗传景观,为进一步的细胞分裂创造了障碍。逆转这个过程可以修复受损的心脏。
生物电相变模式决定了脊椎动物的第一次心跳
A bioelectrical phase transition patterns the first vertebrate heartbeats. Nature
挺有趣的科学问题
- 规律的心跳对于脊椎动物的生命至关重要。在成熟的心脏中,该功能由解剖学上定位的起搏器驱动。相比之下,起搏能力广泛分布在早期胚胎心脏中,这就提出了组织尺度活动如何在胚胎发育过程中首先建立然后维持的问题。心脏从无声到跳动的最初转变从未在单个电事件的时间尺度上得到表征,并且早期心跳的空间和时间结构仍然知之甚少。
- 利用全光学电生理学,我们捕获了斑马鱼的第一次心跳,并分析了围绕这一奇异事件的心脏兴奋性和传导的发展。最初的几次心跳突然出现,具有不规则的心跳间隔,在原始心脏中连贯地传播,并且从不同动物和随时间变化的基因座发出。通过 CaV1.2 驱动的动作电位上冲程不变圆分叉上的噪声鞍节点很好地描述了生物电动力学。
- 我们的工作展示了单细胞生物电特性的逐步且很大程度上异步的发展如何产生从静止到协调跳动的定型且稳健的组织尺度转变。
转座子编码的核酸酶使用引导RNA来促进其自私传播
Transposon-encoded nucleases use guide RNAs to promote their selfish spread. Nature
- 插入序列是细菌中发现的紧凑且普遍的转座元件,仅编码其动员和维持所需的基因。 IS200 和 IS605 家族转座子经历 TnpA 转座酶催化的“剥离和粘贴”转座,但它们也编码多种 TnpB 和 IscB 家族蛋白,这些蛋白在进化上分别与 CRISPR 相关效应子 Cas12 和 Cas9 相关。最近的研究表明,TnpB 和 IscB 作为 RNA 引导的 DNA 核酸内切酶发挥作用,但这种活性的更广泛的生物学作用仍然是个谜。
- 在这里,我们证明 TnpB 和 IscB 对于防止 TnpA 转座机制导致的永久性转座子丢失至关重要。我们从嗜热脂肪地芽孢杆菌中选择了一个相关插入序列家族,它们编码多个 TnpB 和 IscB 直系同源物,并表明单个 TnpA 转座酶对转座子动员具有广泛的活性。转座子侧翼序列重新连接后形成的供体接头被 RNA 引导的 TnpB 和 IscB 核酸酶有效地靶向切割,并且与单独表达 TnpA 的条件相比,TnpB 和 TnpA 的共表达导致转座子保留显着增加。
- 值得注意的是,TnpA 和 TnpB 还刺激了重组频率,超过了单独使用 TnpB 时观察到的重组频率。
- 总的来说,这项研究揭示了 RNA 引导的 DNA 切割作为一种原始生化活动而出现,以偏向转座元件的自私遗传和传播,后来在用于抗病毒防御的 CRISPR-Cas 适应性免疫的进化过程中被采用。
独特保存的肠道内容物阐明了三叶虫的古生理学
Uniquely preserved gut contents illuminate trilobite palaeophysiology. Nature
- 三叶虫是最具标志性的化石之一,在从寒武纪早期到二叠纪末期的 2.7 亿年历史的大部分时间里,它们构成了海洋生态系统的重要组成部分。迄今为止,已描述了超过 20,000 个物种,推测它们的生活方式从动物穴居到水柱中的浮游生活。推断的营养作用范围从食腐动物到捕食者,但所有这些都是基于间接证据,例如身体和肠道形态、保存模式和归因的摄食痕迹;尚未描述过具有内部肠道内容物的三叶虫标本。
- 在这里,我们展示了奥陶纪三叶虫 Bohemolichas incola 完整且详细的肠道内容物,其三维保存在硅质结节中,并通过同步加速器显微断层扫描进行可视化。紧密堆积、几乎连续的肠道填充物由部分破碎的钙质贝壳组成,表明进食强度很高。壳不溶解意味着整个肠道的中性或碱性环境支持消化酶,与现代甲壳类动物或螯合物中的消化酶相当。
- 钻入三叶虫尸体的食腐动物以眉间下方的软组织为目标,但避开了肠道,这表明存在有害条件,并且可能存在持续的酶活性。
营养胁迫下的蛋白质组普查揭示了高尔基噬膜受体
Proteome census upon nutrient stress reveals Golgiphagy membrane receptors. Nature
- 在营养应激期间,巨自噬会降解细胞大分子,从而提供生物合成构件,同时重塑蛋白质组。虽然负责启动巨自噬的机制已得到很好的表征,但我们对单个蛋白质、蛋白质复合物和细胞器被选择用于自噬降解的程度以及潜在的靶向机制的理解是有限的。
- 在这里,我们使用正交蛋白质组策略来提供哺乳动物细胞营养应激期间自噬货物的空间蛋白质组普查。我们发现巨自噬对膜结合细胞器(主要是高尔基体和内质网)的回收具有选择性。
- 通过自噬货物优先级排序,我们确定了膜嵌入蛋白 YIPF3 和 YIPF4 的复合物作为高尔基体吞噬的受体。在营养胁迫期间,YIPF3 和 YIPF4 通过 LIR 基序与 ATG8 相互作用,并被动员到自噬体中,该自噬体在需要规范自噬机制的过程中运输到溶酶体。缺乏 YIPF3 或 YIPF4 的细胞在营养应激期间选择性地消除特定的高尔基体膜蛋白群。此外,YIPF3/4 在体外干细胞向神经元谱系的程序性转化过程中的高尔基体重塑中发挥着类似的作用。
- 总的来说,这项研究揭示了营养应激依赖性蛋白质组重塑过程中膜蛋白货物的优先顺序,并确定了一个意想不到的需要膜嵌入受体的高尔基体重塑途径。
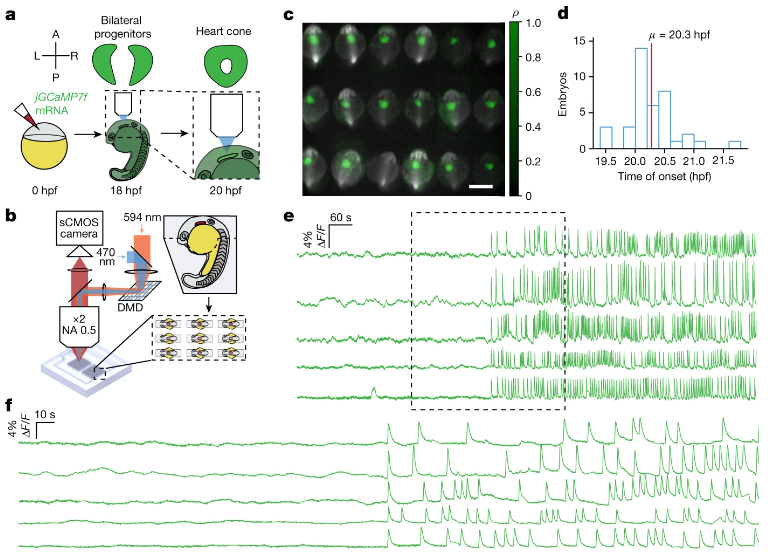
接种不同剂量灭活病毒疫苗的 Omicron 感染受试者的系统免疫分析
上海交通大学医学院附属瑞金医院呼吸与危重症医学科
- 基于 SARS-CoV-2 主要毒株的疫苗接种以加强依赖性方式对 Omicron 变体引发的感染、症状发生和疾病严重程度发挥保护作用。然而,其根本机制仍不清楚。
- 在 2022 年上海 Omicron 疫情期间,我们招募了 122 名感染成人和 50 名未接种疫苗或接种过两剂或三剂 COVID-19 灭活疫苗的未感染对照者,并对 41 重 CyTOF、RNA-seq 和 Olink 进行了综合分析他们的外周血样本。
- 加强疫苗后HLA-DRhi经典单核细胞、非经典单核细胞和Th1样Tem的频率呈增加趋势,而Treg的频率则降低,并且它们以疫苗剂量依赖性方式影响症状的发生。
- 相互关联和机制分析表明,加强疫苗接种诱导了单核细胞训练,这将促进单核细胞激活和成熟,而不是在 Omicron 感染时分化为骨髓源性抑制细胞。
- 总体而言,我们的研究提供了关于加强疫苗接种如何阐述针对 SARS-CoV-2 变体的保护性免疫的见解。
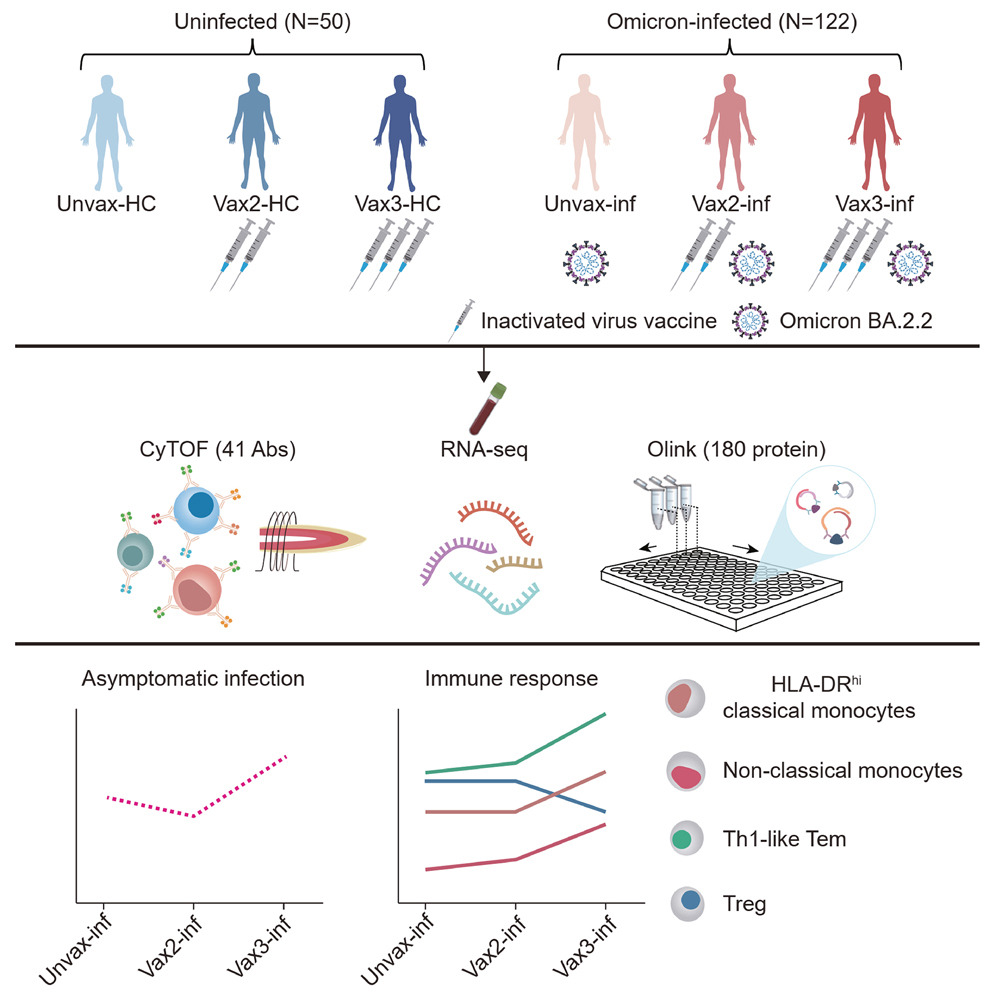
局部微环境驱动人脑肿瘤中性粒细胞的激活
The local microenvironment drives activation of neutrophils in human brain tumors. Cell
似乎并没有很新的发现
- 中性粒细胞是循环系统中丰富的免疫细胞,经常大量浸润肿瘤。然而,它们在不同癌症类型中的精确功能仍不完全清楚,包括在大脑微环境中。
- 因此,我们研究了神经胶质瘤和脑转移患者肿瘤组织中的中性粒细胞以及匹配的外周血,并在此描述了对这些组织中中性粒细胞表型和功能的首次深入分析。
- 人类和小鼠的正交分析策略表明,脑肿瘤相关中性粒细胞(TAN)与血液中性粒细胞显着不同,并且具有延长的寿命以及免疫抑制和促血管生成的能力。 TAN 表现出独特的炎症特征,由肿瘤坏死因子 α (TNF-ɑ) 和铜蓝蛋白等可溶性炎症介质的组合驱动,与神经胶质瘤相比,脑转移瘤中的 TAN 更明显。
- 骨髓细胞,包括肿瘤相关巨噬细胞,出现在促炎介质网络的核心,支持调节人脑肿瘤整体免疫抑制的关键骨髓生态位的概念。
性别特异性因子 SOA 控制按蚊的剂量补偿
The sex-specific factor SOA controls dosage compensation in Anopheles mosquitos. Nature
- 按蚊是性别差异在其生物学中发挥核心作用的数千种蚊子之一,因为只有雌性需要吸血才能产卵。性别分化受性染色体调节,但它们的存在会造成男性 (XY) 和女性 (XX) 之间的剂量不平衡。剂量补偿(DC)可以重新平衡性染色体基因的表达,但由于 DC 机制仅在少数模式生物中得到充分表征,因此有关其进化多样性和功能必要性的关键问题仍未解决 。
- 在此我们报告这一发现以前未表征的基因(SOA,用于性染色体激活)作为疟疾蚊子冈比亚按蚊 DC 的主要调节因子。性别特异性选择性剪接可阻止女性 SOA 蛋白的功能表达。雄性亚型编码一种 DNA 结合蛋白,可结合活性 X 染色体基因的启动子。表达雄性 SOA 足以在雌性细胞中诱导 DC。缺乏 SOA 的雄性蚊子或异位表达雄性亚型的雌性蚊子表现出 X 染色体失调,这与生存能力相容,但会导致发育迟缓。
- 因此,我们对非模型生物体中第一个 DC 主调节因子的分子分析阐明了导致建立染色体特异性微调机制的进化步骤。
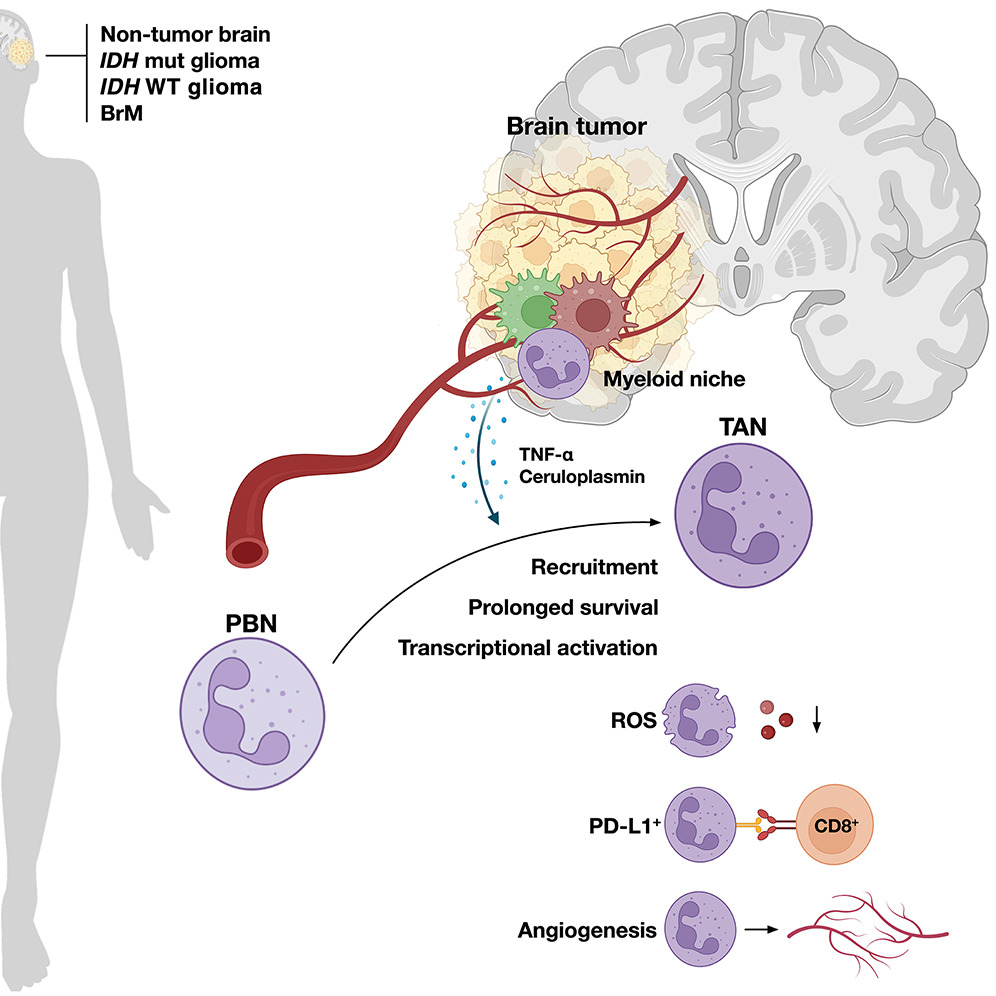
合成 Par 极性诱导未极化哺乳动物细胞的细胞骨架不对称
Synthetic Par polarity induces cytoskeleton asymmetry in unpolarized mammalian cells. Cell
- 极化细胞依靠极化细胞骨架来发挥作用。然而,皮质极性线索如何诱导细胞骨架极化仍然难以捉摸。
- 在这里,我们利用最近建立的设计的二维蛋白质阵列来异位设计几乎任何感兴趣的蛋白质在各种细胞类型的有丝分裂过程中的皮质极性。这使得能够直接操纵极性信号传导并识别足以细胞骨架极化的皮质线索。
- 使用这种测定,我们剖析了 Par 复合体途径的逻辑,Par 复合体途径是不对称细胞分裂过程中细胞骨架极性的关键调节因子。我们表明,任何 Par 复合体亚基的皮质聚类足以触发复合体组装,并且复合体组装的主要动力学障碍是 Par6 自抑制的缓解。
- 此外,我们发现诱导皮质 Par 复合体极性会诱导非极化哺乳动物细胞中不对称细胞分裂的两个特征:纺锤体方向(通过 Par3 发生)和中央纺锤体不对称(取决于 aPKC 活性)。
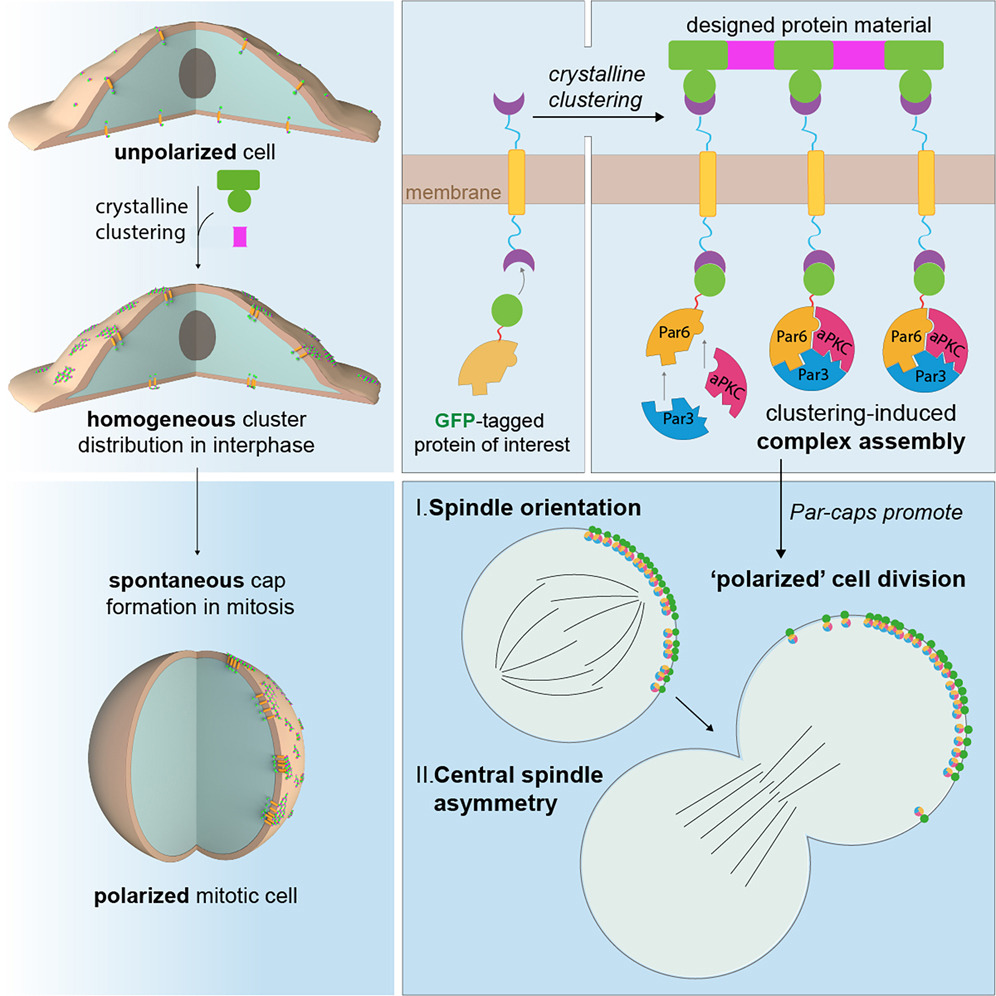
心脏疤痕组织中的成纤维细胞直接调节心脏兴奋性和心律失常发生
Fibroblasts in heart scar tissue directly regulate cardiac excitability and arrhythmogenesis. Science
实验方法挺巧秒。实验分组中必须要有未发生心肌梗塞的转基因小鼠!
- 心脏损伤后,死亡的心肌被疤痕组织取代。成纤维细胞可以与肌细胞电耦合,成纤维细胞膜电位的变化可以导致肌细胞兴奋性,这表明疤痕组织中的成纤维细胞-肌细胞耦合可能是心律失常发生的原因。然而,肌细胞和成纤维细胞电耦合的生理相关性及其对体内心脏兴奋性的影响尚未得到证实。
- 我们对小鼠进行了基因改造,使其仅在心脏成纤维细胞中表达光遗传学阳离子通道 ChR2 (H134R)。心肌梗塞后,疤痕组织的光刺激引起器官范围的心脏兴奋并诱发这些动物的心律失常。
- 通过实验方法补充计算模型,我们发现间隙连接和触觉耦合以协同但功能冗余的方式兴奋与成纤维细胞耦合的肌细胞。
植物的表观遗传进化时钟
An evolutionary epigenetic clock in plants. Science
挺有意思。该分子钟与物种进化间是否存在因果关系?
- 分子钟是在宏观进化时间尺度(约 105 至 108 年)内测定谱系之间差异的基础。然而,基于 DNA 的经典时钟走得太慢,无法告诉我们最近的过去。
- 在这里,我们证明植物基因组中胞嘧啶子集的随机 DNA 甲基化变化表现出类似时钟的行为。这种“后突变时钟”比基于 DNA 的时钟快几个数量级,可以进行数年至数百年规模的系统发育探索。我们通过实验证明,表突变时钟概括了自花受精植物拟南芥和克隆海草大叶藻中种内系统发育树的已知拓扑结构和分支时间,它们代表了植物繁殖的两种主要模式。
- 这一发现将为植物生物多样性的高分辨率时间研究开辟新的可能性。
控制新陈代谢和食物摄入的整合神经回路
Integrative neurocircuits that control metabolism and food intake. Science
这是用新的技术揭示了一个已被研究清楚的神经回路?
- 越来越多的超重和肥胖人群表现出罹患多种肥胖相关疾病的倾向,例如 2 型糖尿病、心血管疾病、某些类型的癌症以及神经退行性疾病。由于能量稳态和外周代谢都是通过大脑协调的,因此迫切需要确定代谢调节的基本神经生物学机制,并确定这些途径的改变如何促进肥胖的发展和肥胖相关代谢紊乱的发生,以便为这些普遍存在的疾病制定治疗干预措施。
- 下丘脑弓状核(ARC)整合多种激素和神经元输入,发出生物体营养可用性的信号。该下丘脑控制系统的核心包括两个神经元群,它们在调节摄食行为、能量消耗和燃料代谢方面发挥几乎相反的功能。刺豚鼠相关肽(AgRP)神经元在能量不足的情况下被激活,被瘦素和胰岛素的燃料通讯信号抑制,并促进觅食和食物消耗。阿黑皮质素原 (POMC) 神经元在正能量平衡状态下被激活,相关的激素变化会减少食物摄入并增加能量消耗。过去 20 年的深入研究表明,这一回路的改变与小鼠模型和人类的肥胖发展存在因果关系。
- 高通量单细胞和单核 RNA 测序方法的最新发展使得能够以前所未有的分子分辨率定义细胞亚群。应用这些技术最近导致下丘脑中许多额外的食物摄入和代谢调节神经元和非神经元细胞群的鉴定。与此同时,功能性分子系统方法不仅可以描述这些新识别的细胞类型在代谢控制中的功能作用,还可以定义神经元网络组织以及评估其在自由行为动物中的活动。这些实验表明,代谢调节神经元在不同的时间尺度上受到调节,包括对食物线索的感觉感知、源自胃肠道的食后信号以及更长期的激素介质。这些信号的整合有助于以稳态方式微调代谢适应和相关行为。这些研究极大地增进了我们对中枢神经系统依赖性代谢控制基本原理的认识。他们还允许定义对抗代谢疾病的新策略。审查中强调了这些最新进展。
- 进一步扩展这些进展将使我们能够更全面地了解啮齿动物模型和人类中保守的代谢调节细胞类型和神经回路。这些新知识将有助于定义它们的放松管制与代谢紊乱的发展之间的关系。此外,此类研究将有助于阐明有前途的新型抗肥胖疗法的作用方式。其中包括胰高血糖素样肽-1 (GLP-1) 受体激动剂以及新开发的针对不同肠源肽受体的多激动剂,临床研究已提供证据证明其在减轻体重和改善代谢方面具有良好的功效。此外,对代谢调节细胞类型的分子特征及其网络相互作用背后的突触机制的更深入了解有可能促进代谢疾病替代治疗策略的发展。
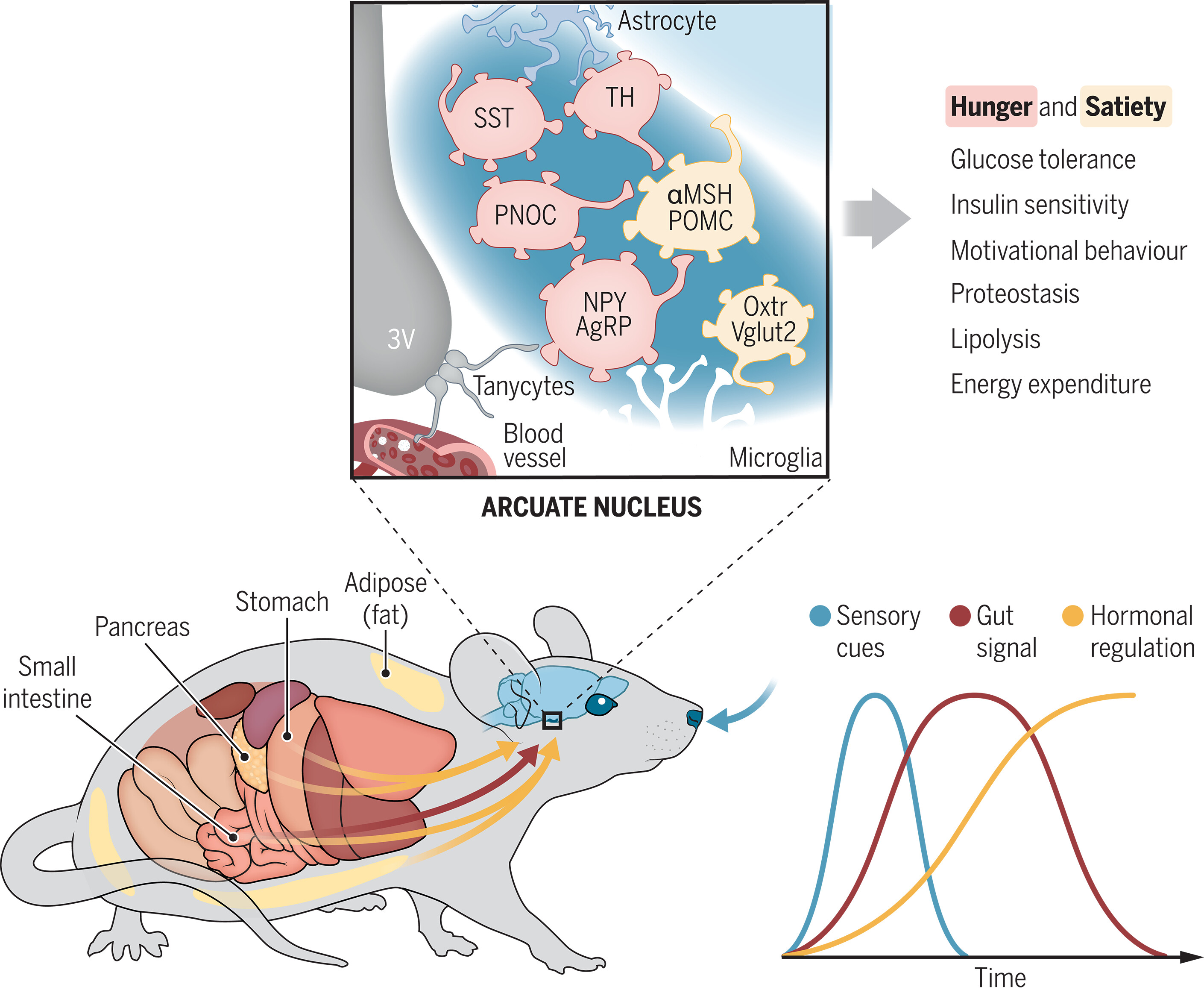
基于 AsCas12f 的紧凑型基因组编辑工具
- SpCas9 和 AsCas12a 被广泛用作人类细胞中的基因组编辑工具。然而,它们相对较大的尺寸限制了货物尺寸限制的腺相关病毒(AAV)载体的递送。来自氧化硫酸杆菌的 V-F Cas12f 型非常紧凑(422 个氨基酸),已被用作紧凑的基因组编辑工具。
- 在这里,我们开发了一种方法,结合深度突变扫描和结构知情设计,成功生成两个 AsCas12f 活性增强 (enAsCas12f) 变体。值得注意的是,enAsCas12f 变体在人类细胞中表现出与 SpCas9 和 AsCas12a 相当的基因组编辑活性。冷冻电子显微镜 (cryo-EM) 结构显示,突变稳定了二聚体的形成,并增强了与核酸的相互作用,从而增强了其 DNA 切割活性。此外,enAsCas12f与伴侣基因一起包装在一体式AAV载体中,在小鼠中表现出有效的敲入/敲除活性和转录激活。
- 总而言之,enAsCas12f 变体可以为体内基因治疗提供最小的基因组编辑平台。
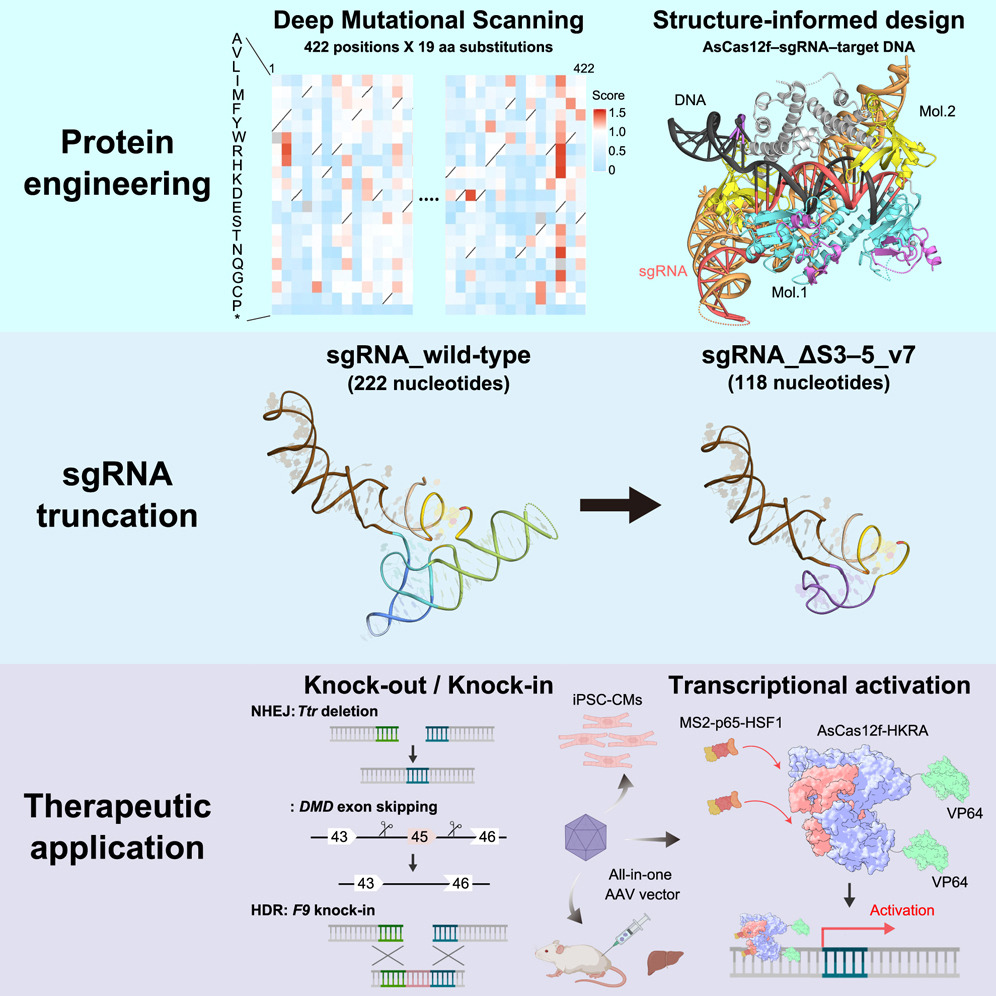
出生后对 SARS-CoV-2 粘膜和全身免疫的多组学分析
Multi-omics analysis of mucosal and systemic immunity to SARS-CoV-2 after birth. Cell
婴儿这种针对病毒的能力的机制是什么?在该现象下,成人免疫是“衰退”了还是“进步”了(或者说这种现象是消极意义多还是积极意义多)?
- 婴儿感染免疫力的动态仍不清楚。在这里,我们采用多组学方法,通过分析之前、期间和之后收集的血液样本和每周鼻拭子,对婴儿和幼儿对 Omicron 和非 Omicron SARS-CoV-2 感染的免疫力进行纵向分析。
- 感染刺激了强大的抗体滴度,与成人不同,这种滴度在长达 300 天内没有显示出衰减的迹象。婴儿产生了强烈的粘膜免疫反应,其特征是炎症细胞因子、干扰素 (IFN) α 和辅助性 T (Th) 17 以及中性粒细胞标记物(白细胞介素 [IL]-17、IL-8 和 CXCL1)。
- 血液中的免疫反应的特点是先天细胞上激活标记物的上调,没有炎症细胞因子,但有几种趋化因子和干扰素α。后者与单细胞多组学测量的骨髓细胞中病毒载量和干扰素刺激基因(ISG)的表达相关。
- 这些数据共同提供了生命最初几周和几个月内对感染的免疫力的概况。
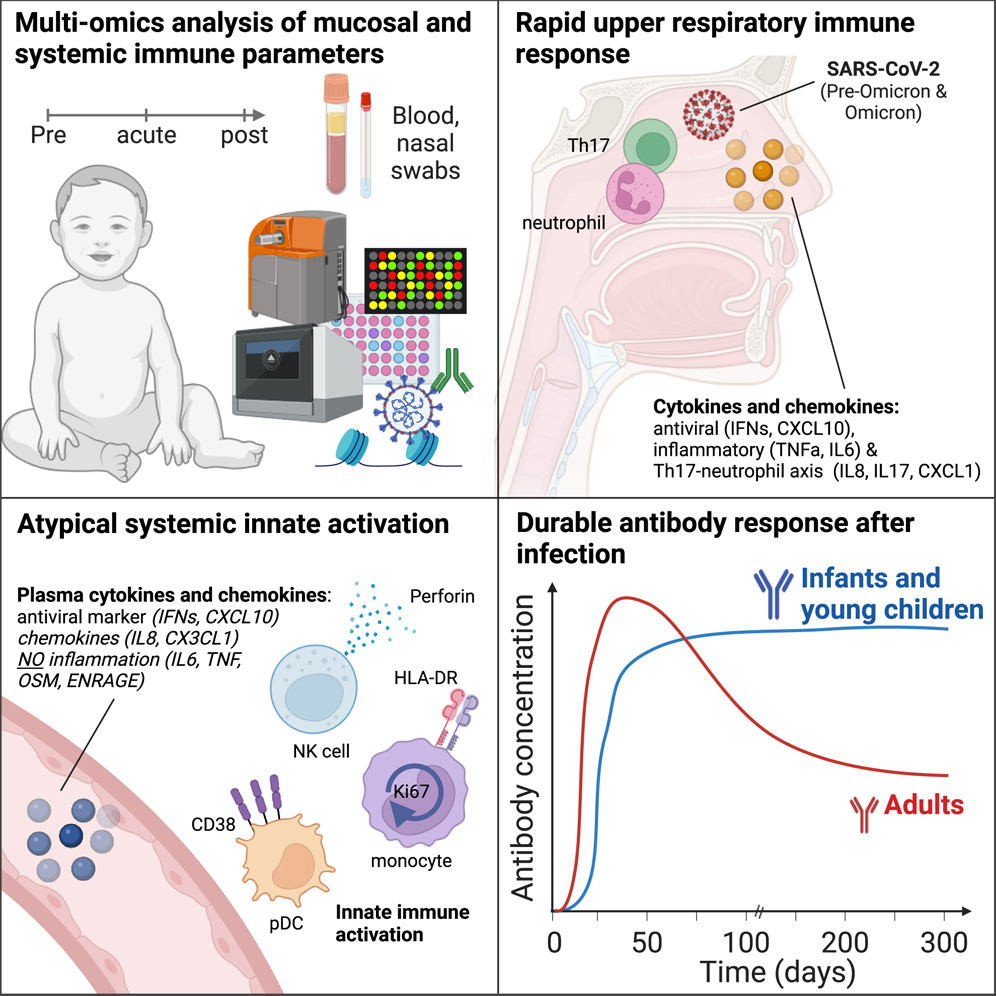
合成敲入序列的模块化汇集发现以编程持久的细胞疗法
Modular pooled discovery of synthetic knockin sequences to program durable cell therapies. Cell
挺特别的研究
- 慢性刺激会导致 T 细胞功能障碍并限制细胞免疫疗法的疗效。需要改进方法来比较大量合成敲入 (KI) 序列以重新编程细胞功能。
- 在这里,我们开发了模块化混合 KI 筛选 (ModPoKI),这是一个使用条形码多顺反子适配器模块化构建 DNA KI 文库的适应性平台。我们构建了两个包含 100 个转录因子 (TF) 和 129 个天然和合成表面受体 (SR) 的 ModPoKI 文库。
- 对不同条件下的人类 TCR 和 CAR-T 细胞进行了超过 30 次 ModPoKI 筛选,发现了一种转录因子 AP4 (TFAP4) 结构,可以增强长期刺激的 CAR-T 细胞的适应性以及体外和体内的抗癌功能。 ModPoKI 的模块化使我们能够生成约 10,000 个成员的 TF 组合库。组合 BATF-TFAP4 多顺反子构建体的非病毒 KI 增强了适应性。过度表达的 BATF 和 TFAP4 共同占据并调节关键基因靶标以重新编程 T 细胞功能。
- ModPoKI 有助于发现复杂的基因结构来编程细胞功能。
肿瘤免疫类
p53 缺失和突变异质性导致具有高肿瘤突变负荷的本地小鼠肺癌模型中的免疫抵抗
美国德克萨斯大学西南医学中心
- 肿瘤突变负荷(TMB)在塑造肿瘤免疫中的作用是一个关键问题,但使用肺癌基因工程小鼠模型(GEMM)尚未解决。为了在肺 GEMM 中诱导 TMB,我们在肺上皮细胞中表达了 DNA 聚合酶-E (POLE)P286R 的超突变变体。
- 将 PoleP286R 等位基因引入 KrasG12D 和 KrasG12D; p53L/L (KP) 模型显着增加其 TMB。 Pole 诱导的免疫原性和对免疫检查点阻断 (ICB) 的敏感性部分依赖于 p53。接受免疫治疗后,肿瘤具有 TP53 截短突变的 NSCLC 患者的生存期比的 TP53WT 患者要短。
- 免疫抵抗部分是由于抗原呈递减少,部分是由于突变异质性。 Pole 突变 KP 肿瘤中总 STING 蛋白水平升高,造成了脆弱性。
- 稳定的多价 STING 激动剂或 p53 诱导剂可提高免疫疗法的敏感性,为这些多克隆肿瘤提供治疗选择。
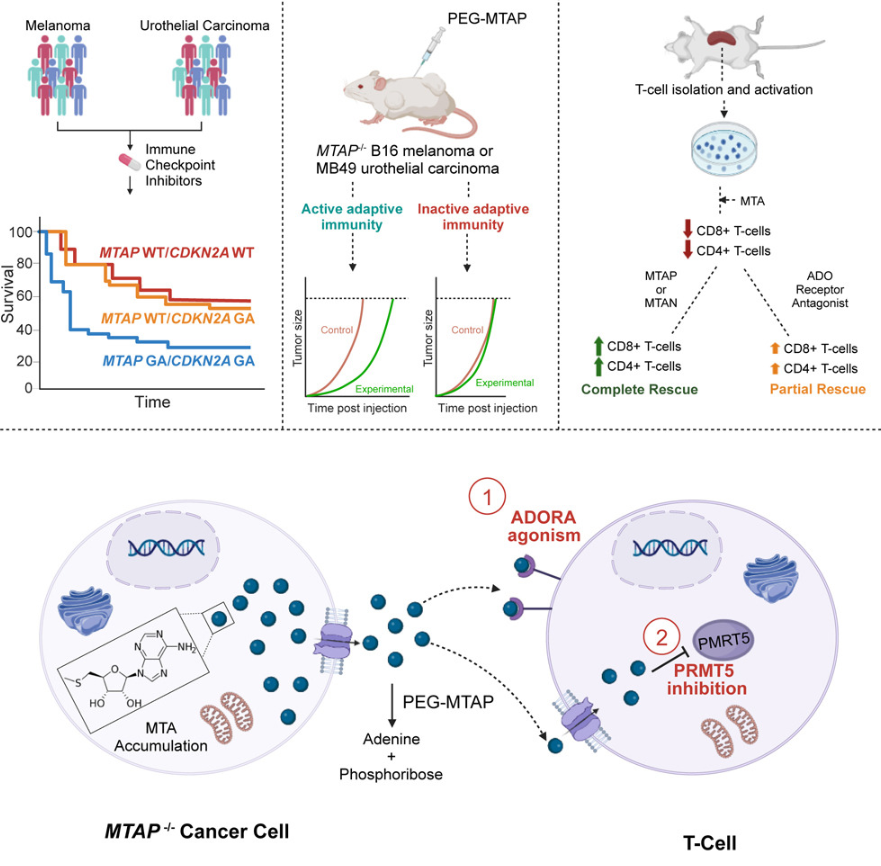
酶介导的甲硫腺苷消耗可恢复 MTAP 缺陷肿瘤中的 T 细胞功能并逆转免疫治疗耐药性
- 含有肿瘤抑制因子 CDKN2A/B 和甲硫腺苷磷酸化酶 (MTAP) 的染色体区域 9p21 是癌症中最常见的基因缺失之一。 9p21 缺失与肿瘤浸润淋巴细胞 (TIL) 减少和免疫检查点抑制剂 (ICI) 治疗耐药相关。以前认为是由 CDKN2A/B 缺失引起的,现在我们发现正是 MTAP 缺失导致 ICI 治疗效果不佳和 TIL 密度降低。
- MTAP 缺失会导致细胞内和细胞外甲硫腺苷 (MTA) 的积累,并通过抑制蛋白精氨酸甲基转移酶 5 (PRMT5) 和腺苷受体激动作用,严重损害 T 细胞功能。施用 MTA 消耗酶可逆转这种免疫抑制作用,增加 TIL 并显着损害肿瘤生长,重要的是,与 ICI 治疗具有良好的协同作用。
- 由于多项研究表明 9p21/MTAP 无效/低的患者存在 ICI 耐药性,因此我们提出 MTA 降解疗法可能通过增强 ICI 有效性而对这些患者产生显着的治疗益处。
抑制性辅助受体 CTLA4 的缺失可增强并激活CAR-T 细胞
- 针对 CD19 的嵌合抗原受体 (CAR) T 细胞疗法在治疗 B 细胞恶性肿瘤方面取得了巨大成功;然而,一些患者由于自体 T 细胞适应性较差而未能做出反应。为了提高缓解率,我们研究了共抑制受体 CTLA4 或 PD-1 的破坏是否可以恢复 CART 功能。
- 在白血病和骨髓瘤的临床前模型中,CRISPR-Cas9 介导的 CTLA4 缺失改善了 CAR T 细胞增殖和抗肿瘤功效。重要的是,这种效应是 CTLA4 特有的,并且在 CAR T 细胞中删除 CTLA4 和/或 PDCD1 后不会出现这种效应。
- 从机制上讲,CTLA4 缺陷使得 CD28 信号传导不受阻碍,并在高抗原负载条件下维持 T 细胞表面的 CAR 表达。在临床研究中,CTLA4 的缺失可以挽救先前 CAR T 细胞治疗失败的白血病患者的 T 细胞功能。
- 因此,选择性删除 CTLA4 可重振功能失调的慢性淋巴细胞白血病 (CLL) 患者 T 细胞,为提高患者对 CAR T 细胞疗法的反应提供策略。
小细胞肺癌细胞和星形胶质细胞之间的串扰模仿大脑发育促进脑转移
Crosstalk between small-cell lung cancer cells and astrocytes mimics brain development to promote brain metastasis. Nat Cell Biol
- 脑转移是小细胞肺癌(SCLC)患者的一个重要临床问题。然而,人们对 SCLC 在大脑中生长的机制仍知之甚少。
- 在这里,通过对小鼠进行颅内注射以及培养中的 SCLC 聚集体和人类皮质类器官之间的组合体,我们发现 SCLC 细胞将反应性星形胶质细胞募集到肿瘤微环境中。 SCLC 细胞和星形胶质细胞之间的这种串扰驱动基因表达程序的诱导,这些基因表达程序类似于神经元和星形胶质细胞早期大脑发育过程中发现的基因表达程序。
- 从机制上讲,SCLC 细胞分泌的大脑发育因子 Reelin 会招募星形胶质细胞至脑转移瘤。这些星形胶质细胞反过来通过分泌 SERPINE1 等神经元促生存因子来促进 SCLC 生长。
- 因此,SCLC 脑转移瘤的生长是通过参与大脑发育过程中神经元-星形胶质细胞相互作用的增选机制来实现的。针对癌症生态系统中激活的此类发育程序可能有助于预防和治疗脑转移。
USP47 抑制 m6A 依赖性 c-Myc 翻译以维持调节性 T 细胞代谢和功能稳态
USP47 inhibits m6A-dependent c-Myc translation to maintain regulatory T cell metabolic and functional homeostasis. J Clin Invest
- Treg 细胞的功能完整性与细胞代谢交织在一起;然而,控制 Treg 细胞代谢程序的机制仍然难以捉摸。
- 在这里,我们发现去泛素酶 USP47 抑制 RNA m6A 阅读器 YTHDF1 介导的 c-Myc 翻译,以维持 Treg 细胞代谢和功能稳态。 USP47 与结直肠癌和胃癌患者样本中的肿瘤浸润 Treg 细胞特征呈正相关。 USP47 消融会损害体内 Treg 细胞的稳态和功能,导致炎症性疾病的发展,并增强抗肿瘤免疫反应。 Treg 细胞中的 USP47 缺陷引发了 c-Myc 蛋白的积累,进而加剧了糖酵解过度。从机制上讲,USP47 阻止 YTHDF1 泛素化,从而减弱 YTHDF1 与翻译起始机制的关联,从而降低基于 m6A 的 c-Myc 翻译效率。
- 我们的研究结果表明,USP47 指导 m6A 依赖性代谢程序来协调 Treg 细胞稳态,并提出了通过靶向 USP47 来选择性调节癌症和自身免疫性疾病的新方法。
NKG2A 是免疫治疗耐药性 MHC-I 异质TNBC中的治疗漏洞
NKG2A is a Therapeutic Vulnerability in Immunotherapy Resistant MHC-I Heterogeneous Triple Negative Breast Cancer. Cancer Discov
- 尽管免疫检查点抑制(ICI)在治疗癌症方面取得了成功,但三阴性乳腺癌(TNBC)患者经常对治疗产生耐药性,其潜在机制尚不清楚。 MHC-I 表达对于抗原呈递和 T 细胞定向免疫治疗反应至关重要。
- 这项研究表明,TNBC 患者在区域 MHC-I 表达方面表现出肿瘤内异质性。在小鼠模型中,MHC-I 的缺失会抵消抗肿瘤免疫和 ICI 反应,而肿瘤内 MHC-I 异质性导致 NK 细胞以 IFNγ 依赖性方式浸润增加。
- 利用空间技术,MHC-I 异质性与抗 PD-L1 治疗的临床耐药性以及人类乳腺肿瘤中 NK:T 细胞比率的增加有关。 MHC-I 异质性肿瘤需要 NKG2A 来抑制 NK 细胞功能。结合抗 NKG2A 和抗 PD-L1 疗法可恢复异质 MHC-I 小鼠模型中的完全反应,这取决于激活的肿瘤浸润 NK 和 CD8+ T 细胞的存在。
- 这些结果表明,类似的策略可能会提高临床试验中患者的利益。
中性粒细胞通过乌头酸脱羧酶1抵抗铁死亡并促进乳腺癌转移
Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab
经典套路
- 转移导致乳腺癌相关死亡。肿瘤浸润性中性粒细胞 (TIN) 会造成免疫抑制并促进转移。 TIN 的治疗衰弱可能会增强免疫治疗,但识别在 TIN 中高表达且功能必需但在肿瘤外中性粒细胞中表达不足的治疗靶点仍然是一个挑战。
- 在这里,使用单细胞 RNA 测序比较小鼠乳腺肿瘤模型中的 TIN 和循环中性粒细胞,我们确定乌头酸脱羧酶 1 (Acod1) 是小鼠 TIN 中上调最多的代谢酶,并验证了人类 TIN 中 Acod1 的高表达。 Acod1 通过 GM-CSF-JAK/STAT5-C/EBPβ 途径激活,产生衣康酸,衣康酸介导 Nrf2 依赖性防御铁死亡并维持 TIN 的持久性。
- Acod1 消融可减少 TIN 浸润,限制转移(但不限制原发肿瘤),增强抗肿瘤 T 细胞免疫,并增强免疫检查点阻断的功效。
- 我们的研究结果揭示了 TIN 如何通过 Acod1 依赖性免疫代谢开关逃避铁死亡,并将 Acod1 作为抵消免疫抑制和改善抗转移免疫治疗的靶点。
临床类
采用观察等待策略管理的直肠癌患者免疫评分活检的国际验证
International Validation of the Immunoscore Biopsy in Patients With Rectal Cancer Managed by a Watch-and-Wait Strategy. J Clin Oncol. full pdf
观察等待策略的具体细节是?
- 目的:目前尚无能够改善通过观察等待 (W&W) 策略管理的直肠癌患者的选择和监测的生物标志物。最近的一项初步研究提出了免疫评分活检 (ISB) 的预后性能。
- 方法:这项国际验证研究纳入了 249 名通过 W&W 策略管理的临床完全缓解 (cCR) 患者。通过数字病理学对治疗前直肠活检中的瘤内 CD3+ 和 CD8+ T 细胞进行定量并转化为 ISB。主要终点是复发时间(TTR;从新辅助治疗结束到局部再生或远处转移日期的时间)。通过针对混杂因素进行调整的分层 Cox 回归分析了 ISB 与结果之间的关联。通过 3’RNA-Seq 和免疫荧光研究了另外 17 名接受新辅助放化疗和手术治疗的患者的肿瘤引流淋巴结 (n = 161) 的免疫状态。
- ISB 高、ISB 中和 ISB 低的 5 年无复发率分别为 91.3% (82.4%-100.0%)、62.5% (53.2%-73.3%) 和 53.1% (42.4%-66.5%)分别(风险比 [HR;低 vs 高],6.51;95% CI,1.99 至 21.28;log-rank P = .0004)。 ISB 还与无病生存显着相关(log-rank P = .0002),并预测局部再生和远处转移。在多变量分析中,ISB 与患者年龄、性别、肿瘤位置、cT 分期(T,原发肿瘤;c,临床)、cN 分期(N,区域淋巴结;c,临床)无关,并且是 TTR 最强的预测因子(HR [ISB 高与低],6.93;95% CI,2.08 至 23.15;P = .0017)。将 ISB 添加到基于临床的模型中显着改善了复发的预测。最后,在 cCR 患者的引流淋巴结中证实了引流淋巴结中的 B 细胞增殖和记忆。
- 结论:ISB 被验证为预测局部再生和远处转移的生物标志物,并逐渐降低贬义结果的风险。
MyPathway 人EGFR2 篮子研究:帕妥珠单抗 + 曲妥珠单抗治疗组织不可知的人表皮生长因子受体 2 改变的晚期实体瘤患者群体
靶向治疗的策略在CUP中同样是成立的
- MyPathway 多篮研究(ClinicalTrials.gov 标识符:NCT02091141)正在评估具有相关分子改变的非适应症肿瘤的靶向治疗。
- 我们在组织不可知的人类表皮生长因子受体 2 (HER2) 扩增和/或过度表达和/或突变实体瘤成人患者队列中评估了帕妥珠单抗 + 曲妥珠单抗。主要终点是客观缓解率(ORR);次要终点包括生存率和安全性。截至数据截止(2022 年 3 月),已有 346 名 HER2 扩增和/或过度表达,伴/不伴 HER2 突变 (n = 263) 或仅 HER2 突变 (n = 83) 的患者接受了治疗。
- HER2 扩增和/或过度表达的患者的 ORR 为 25.9%(68/263,95% CI,20.7 至 31.6),其中包括 5 例完全缓解(尿路上皮 [n = 2]、唾液腺 [n = 2] 和结肠) [n = 1] 癌症)。野生型 (ORR, 28.1%) 的活性高于突变型 KRAS (ORR, 7.1%)。在 HER2 扩增的患者中,免疫组织化学 (IHC) 3+ (41.0%; 32/78) 或 2+ (21.9%; 7/32) 患者的 ORR 数值高于 1+ (8.3%; 1/12) 患者或无表达(0%;0/20)。在仅有 HER2 突变的患者中,ORR 为 6.0%(5/83,95% CI,2.0 至 13.5)。
- 帕妥珠单抗 + 曲妥珠单抗在具有野生型 KRAS 的各种 HER2 扩增和/或过表达肿瘤中显示出活性,活性范围取决于肿瘤类型,但在 KRAS 突变、单独 HER2 突变或 0-1+ HER2表达的情况下活性有限。
吉西他滨+顺铂+纳武单抗作为肌肉浸润性膀胱癌的器官保留治疗颇有潜力
- 膀胱切除术是肌层浸润性膀胱癌 (MIBC) 的标准治疗方法,但它会改变生活。我们启动了一项 2 期研究,其中 MIBC 患者接受四个周期的吉西他滨、顺铂加纳武单抗治疗,然后进行临床再分期。达到临床完全缓解(cCR)的患者无需进行膀胱切除术即可继续治疗。共同主要目标是评估 cCR 率和 cCR 对复合结果的阳性预测价值:放弃立即膀胱切除术的患者的 2 年无转移生存率或选择立即膀胱切除术的患者的 <ypT1N0。
- 共有 76 名患者入组;其中,33 人实现了 cCR(43%,95% 置信区间 (CI):32%-55%),实现 cCR 的 33 人中有 32 人选择放弃立即膀胱切除术。 cCR 的阳性预测值为 0.97(95% CI:0.91,1),满足共同主要目标。
- 最常见的不良事件是疲劳、贫血、中性粒细胞减少和恶心。预先指定基因(ATM、RB1、FANCC 和 ERCC2)的体细胞改变或肿瘤突变负荷增加并不能提高 cCR 的阳性预测值。外周血质量细胞计数和可溶性蛋白分析物的探索性分析证明了基线和治疗中免疫环境与临床结果之间的关联。
- 吉西他滨、顺铂加纳武单抗后严格定义的 cCR 促进了膀胱保留,值得进一步研究。 ClinicalTrials.gov 标识符:NCT03451331。
Olaparib+abiraterone vs. placebo+abiraterone对未通过同源重组修复突变状态选择的转移性去势抵抗性前列腺癌患者无显著获益
- PROpel 达到了其主要终点,显示对于未通过同源重组修复突变 (HRRm) 状态选择的一线转移性去势抵抗性前列腺癌 (mCRPC) 患者,奥拉帕尼加阿比特龙与安慰剂加阿比特龙相比,放射学无进展生存期有统计学显着改善,在所有预先指定的亚组中观察到益处。在这里,我们报告最终的预先指定的总体生存分析。
- 这是一项随机、双盲 3 期试验,在全球 17 个国家的 126 个中心进行。年龄至少 18 岁、东部肿瘤合作组表现状态 0-1、预期寿命至少 6 个月、既往未接受过 mCRPC 全身治疗且未按 HRRm 状态选择的 mCRPC 患者被随机分配 (1:1),由一种交互式语音应答系统-交互式网络应答系统,醋酸阿比特龙(口服,1000毫克,每天一次)加泼尼松或泼尼松龙联合奥拉帕尼(口服,300毫克,每天两次)或安慰剂。患者、研究者和研究中心工作人员对药物分配情况不知情。分层因素是转移部位和转移性激素敏感癌症阶段之前使用的多西紫杉醇。放射学无进展生存期是主要终点,总生存期是 α 对照(预先指定最终分析的 α 阈值:0·0377 [双侧])的关键次要终点,在意向治疗人群中进行评估。对所有接受至少一剂研究药物的患者进行安全性评估。该研究已在 ClinicalTrials.gov 注册,NCT03732820,并已完成且不再招募。
- 2018年10月31日至2020年3月11日期间,对1103名患者进行了筛查,其中399名患者被随机分配至奥拉帕尼加阿比特龙治疗组,397名患者被随机分配至安慰剂加阿比特龙组。经审查数据的患者总生存期中位随访时间,奥拉帕尼加阿比特龙组为 36·6 个月 (IQR 34·1-40·3),安慰剂加阿比特龙组为 36·5 个月 (33·8-40·3)。奥拉帕尼加阿比特龙组的中位总生存期为 42·1 个月(95% CI 38·4-未达到),安慰剂加阿比特龙组的中位总生存期为 34·7 个月(31·0-39·3)(风险比 0·81, 95% CI 0·67-1·00;p=0·054)。
- 最常见的 3-4 级不良事件是贫血,奥拉帕尼加阿比特龙组 398 名患者中有 64 名患者 (16%) 报告,安慰剂加阿比特龙组 396 名患者中有 13 名患者 (3%) 报告。奥拉帕尼加阿比特龙组报告了 161 例(40%)严重不良事件,安慰剂加阿比特龙组报告了 126 例(32%)发生严重不良事件。安慰剂加阿比特龙组中有 1 例因间质性肺疾病死亡,被认为与治疗相关。
- 总之,在最终的预设分析中,治疗组之间的总生存率没有显着差异。
其它类
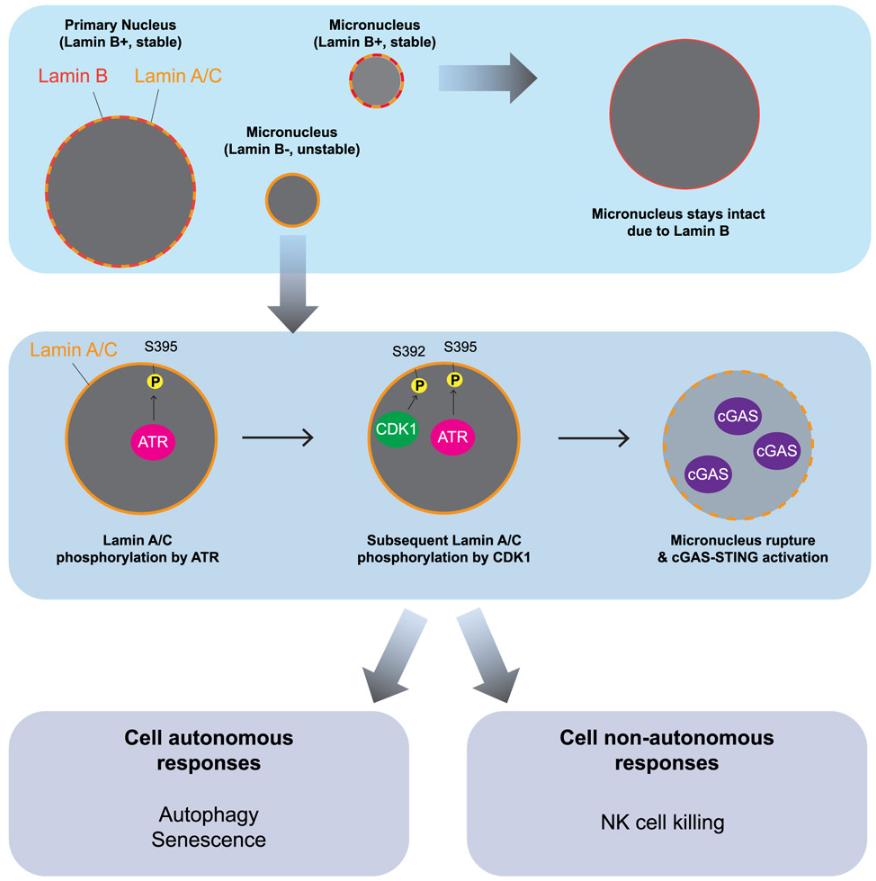
ATR 通过破坏微核促进受损 DNA 和受损细胞的清除
ATR promotes clearance of damaged DNA and damaged cells by rupturing micronuclei. Mol Cell
- 人类共济失调毛细血管扩张症突变和 Rad3 相关 (ATR) 激酶在细胞核中发挥作用,以保护基因组完整性。微核(micronuclei, MN)由基因组和染色体不稳定引起,并导致非整倍体和染色体碎裂,但人们对微核如何去除知之甚少。
- 在这里,我们发现 ATR 在 MN 中具有活性,并通过磷酸化 Lamin A/C Ser395 来促进其在 S 期的破裂,从而引发 Ser392 进行 CDK1 磷酸化并使 MN 包膜不稳定。在含有 MN 的细胞中,抑制 ATR 或 CDK1 可减少 MN 破裂。因此,ATR 抑制剂 (ATRi) 会减少细胞质 DNA 传感器 cGAS 的激活,并损害 MN 中 cGAS 依赖性自噬体的积累和微核 DNA 的清除。此外,ATRi 可减少 cGAS 介导的衰老和自然杀伤细胞对携带 MN 的癌细胞的杀伤。
- 因此,除了典型的 ATR 信号通路外,ATR-CDK1-Lamin A/C 轴还可促进 MN 破裂,以清除受损的 DNA 和细胞,通过意想不到的细胞自主和细胞非自主机制保护细胞群中的基因组。
以单细胞水平分辨率识别细胞粘附或侵入细菌以及相关的宿主转录组
- 单细胞 RNA 测序 (scRNAseq) 技术有助于揭示和描述哺乳动物组织(包括实体瘤)内的细胞异质性。然而,许多这些技术都应用RNA的poly(A)选择,因此主要集中于确定肿瘤微环境的真核细胞成分的基因特征。微生物组分析揭示了主要癌症类型的人类肿瘤组织中存在微生物生态系统,包括细菌和真菌。成像数据显示肿瘤内细菌可能位于上皮细胞和免疫细胞类型内。然而,由于细菌 RNA 通常缺乏 Poly(A) 尾,标准 scRNAseq 方法捕获肿瘤微环境中这种微生物成分的能力有限。
- 为了克服这个问题,我们描述了入侵粘附定向表达测序 (INVADEseq) 方法,除标准引物外,我们还通过引入针对细菌 16S 核糖体 RNA 基因保守区域的引物来适应 10x Genomics 5′ scRNAseq 方案用于真核聚 (A) RNA 选择。这种“附加”方法能够以真核单细胞水平分辨率生成真核和细菌 DNA 文库,利用 10x 条形码识别单细胞与胞内细菌。 INVADEseq方法需要30小时才能完成,包括组织处理、测序和计算分析。
- 作为一项成果,INVADEseq 已被证明是人类癌细胞系和患者肿瘤样本中的可靠工具,通过检测含有细菌的人类细胞的比例以及人类细胞和细胞内细菌的身份,以及识别受调节的宿主转录程序基于相关细菌。
(~ ̄▽ ̄)~ 光免疫技术:基于分子的靶细胞和微生物消除
了解一下基本原理,似乎挺有前景
- 微生物病原体,包括细菌、真菌和病毒,可以对临床使用的药物产生耐药性;因此,寻找新的治疗药物是一个持续的挑战。最近,我们报道了光免疫抗菌策略(photoimmuno-antimicrobial strategy, PIAS),这是一种光免疫技术,能够从分子角度靶向消除多种微生物,包括病毒病原体严重急性呼吸综合征冠状病毒和多重耐药细菌病原体耐甲氧西林金黄色葡萄球菌 (MRSA)。
- PIAS 的工作原理与光免疫疗法 (photoimmunotherapy, PIT) 相同,自 2020 年以来,光免疫疗法已在日本用于治疗复发性头颈癌。PIAS 和 PIT 均使用与酞菁衍生物染料缀合的单克隆抗体,该染料在光激活时会发生形状变化。这种形状变化会引起抗体-染料缀合物的结构变化,从而导致缀合物的结合位点内产生物理应力并破坏它们。因此,靶向准确性和灵活性可以根据所用抗体的特异性来确定。
- 在本协议中,我们描述了如何设计治疗策略、用染料标记单克隆抗体并表征产品。我们提供了如何在体外和体内设置和执行 PIAS 和 PIT 应用程序的详细示例。这些例子包括使用MRSA作为代表性受试者针对微生物的PIAS、在VeroE6/TMPRSS2细胞中使用严重急性呼吸综合征冠状病毒2针对病毒的PIAS、针对MRSA感染动物的PIAS以及针对癌细胞的体外和体内PIT。体外和体内方案可分别在约 3 小时和 2 周内完成。
大斯托克斯位移荧光 RNA 用于活细胞中的双发射荧光和生物发光成像
Large Stokes shift fluorescent RNAs for dual-emission fluorescence and bioluminescence imaging in live cells. Nat Methods
中国-华东理工大学-光遗传学与合成生物学跨学科研究中心-生物反应器工程国家重点实验室
- 荧光 RNA,即结合并激活小荧光染料的适体,为活细胞中 RNA 的可视化提供了一种特别有吸引力的方法。然而,由于缺乏具有生物正交性和合适光谱特性的明亮且稳定的荧光RNA,多种RNA的同时成像仍然具有挑战性。
- 在这里,我们开发了君子兰,这是一系列小型、单体且稳定的橙色至红色荧光 RNA,斯托克斯位移高达 108 nm,能够对 RNA 进行简单而稳健的成像,同时对目标 RNA 的定位和功能的干扰最小。与 Pepper 荧光 RNA 结合,君子兰能够对细胞 RNA 和基因组位点进行单激发双发射双色成像。君子兰还可以通过活细胞和体内的生物发光成像来检测 RNA-蛋白质相互作用。
- 我们相信,这些大斯托克斯位移荧光 RNA 将成为跟踪和量化不同生物过程中多种 RNA 的有用工具。
Pleconaril 和利巴韦林治疗新发 1 型糖尿病:一项 2 期随机试验
Pleconaril and ribavirin in new-onset type 1 diabetes: a phase 2 randomized trial. Nat Med
- 先前的研究表明,新诊断的 1 型糖尿病 (T1D) 患者的胰岛存在低度肠道病毒感染。在糖尿病病毒检测 (DiViD) 干预中,一项 2 期、安慰剂对照、随机、平行组、双盲试验中,96 名新发 T1D 儿童和青少年(6-15 岁)接受了普乐康尼和利巴韦林 (n = 47) 或安慰剂 (n = 49) 6 个月,目的是保留 β 细胞功能。主要终点是使用混合线性模型开始治疗后 12 个月(诊断后不到 3 周)的平均刺激 C 肽曲线下面积 (AUC)。该模型使用基线、3 个月、6 个月和 1 年时的纵向对数转换血清 C 肽 AUC。
- 12 个月时,与安慰剂组相比,普乐那利和利巴韦林治疗组的血清 C 肽 AUC 达到主要终点(线性混合模型中平均边际效应 = 0.057;95% 置信区间 = 0.004-0.11, P = 0.037)。治疗耐受性良好。结果表明,抗病毒治疗可以保留新发 T1D 儿童和青少年的残余胰岛素分泌。
- 这为进一步评估预防和治疗 T1D 的抗病毒策略提供了依据。欧盟药物监管机构临床试验标识符:2015-003350-41。
亚细胞 K+ 成像的DNA纳米装置
Detecting organelle-specific activity of potassium channels with a DNA nanodevice. Nat Biotechnol
美国芝加哥大学化学系
系列研究
- 细胞表面钾离子 (K+) 通道通过控制 K+ 渗透性来调节营养物质运输、细胞迁移和细胞间通讯,并且被认为仅在质膜上活跃。尽管这些通道穿过高尔基体网络、早期内体和再循环内体,但它们在这些细胞器中是否活跃尚不清楚。
- 在这里,我们描述了一种称为 pHlicKer 的 pH 可校正、比例式 K+ 报告基因,用它来探测原型电压门控 K+ 通道 Kv11.1 的区室特异性活性,并表明该细胞表面通道在细胞器中具有活性。在表达野生型 Kv11.1 通道的细胞中,细胞器中的管腔 K+ 增加,但在使用当前阻断剂治疗后则没有增加。运输功能受损的突变 Kv11.1 通道无法增加再循环内体中的 K+ 水平,这一效应可通过药理学纠正来挽救。
- 通过提供一种绘制 K+ 通道细胞器特异性活性的方法,pHlicKer 技术可以帮助识别新的细胞器 K+ 通道或具有细微功能的通道调节剂。
亚细胞 Na+ 成像的DNA纳米装置
A DNA nanodevice for mapping sodium at single-organelle resolution. Nat Biotechnol
美国芝加哥大学化学系
系列研究
- 细胞钠离子 (Na+) 稳态是生物体生理学不可或缺的一部分。我们目前对 Na+ 稳态的理解很大程度上局限于质膜上的 Na+ 转运。细胞器也可能有助于 Na+ 稳态;然而,Na+ 穿过细胞器膜的方向未知,因为细胞器 Na+ 无法成像。
- 在这里,我们报道了一种不依赖于 pH 值、可靶向细胞器的比例探针,可报告腔内 Na+。它是一种 DNA 纳米装置,含有 Na+ 敏感荧光团、参考染料和细胞器靶向结构域。
- 通过测量哺乳动物细胞和秀丽隐杆线虫中单内体分辨率的 Na+,我们发现内溶酶体途径的每个阶段的腔内 Na+ 水平超过胞质水平,并随着内体成熟而降低。此外,我们发现线虫中溶酶体 Na+ 水平受到 Na+/H+ 交换剂 NHX-5 的调节,以响应盐胁迫。
- 亚细胞 Na+ 成像的能力将在更高的细胞细节水平上揭示 Na+ 稳态机制。
针对成人弥漫性中线神经胶质瘤的 H3K27M 靶向疫苗
A H3K27M-targeted vaccine in adults with diffuse midline glioma. Nat Med
在某些比较前沿的研究中,“based on physician’s discretion”都可以是依据之一嘛 (ฅ´ω`ฅ)
- 将组蛋白 H3 (H3K27M) 中的第 27 位赖氨酸替换为蛋氨酸,定义了弥漫性神经胶质瘤的侵袭性亚型。先前的研究表明,H3K27M 特异性长肽疫苗 (H3K27M-vac) 可诱导突变特异性免疫反应,从而控制主要组织相容性复合物人源化小鼠中的 H3K27M+ 肿瘤。
- 在这里,我们描述了在知情使用的基础上使用 H3K27M-vac 对 8 名患有进展性 H3K27M+ 弥漫性中线神经胶质瘤的成年患者进行的首次人体治疗。 5 名患者根据医生的判断接受了 H3K27M-vac 联合抗 PD-1 治疗。重复接种 H3K27M-vac 疫苗是安全的,并且在 8 名患者中的 5 名中诱导了 CD4+ T 细胞主导的突变特异性免疫反应,涉及多种人类白细胞抗原类型。接种疫苗后中位无进展生存期为 6.2 个月,中位总生存期为 12.8 个月。一名 H3K27M-vac 后出现强烈突变特异性 T 细胞反应的患者出现假性进展,随后持续完全缓解超过 31 个月。
- 我们的数据证明了 H3K27M-vac 在进行性 H3K27M+ 弥漫性中线胶质瘤患者中的安全性和免疫原性。
EzMechanism:提出酶反应催化机制的自动化工具
EzMechanism: an automated tool to propose catalytic mechanisms of enzyme reactions. Nat Methods
- 多年来,人们利用实验和模拟方法研究了数百种酶反应机制。这些关于生物催化的丰富文献现在已经成熟,可以用作研究酶机制的新的基于知识的方法的基础。
- 在这里,我们提出了一种工具,能够根据从机制和催化位点图谱(酶机制数据库)编制的一组催化规则,自动推断给定三维活性位点和酶反应的机制路径。
- 每个人都可以通过网络用户界面使用 EzMechanism(发音为“Easy” Mechanism)。在研究机制时,EzMechanism 通过确保考虑相关信息(源自相关和不相关酶的文献)来促进和改进假设的生成。
- 我们在一组 62 种酶上验证了 EzMechanism,并确定了进一步改进的途径,包括需要额外的和更通用的催化规则。
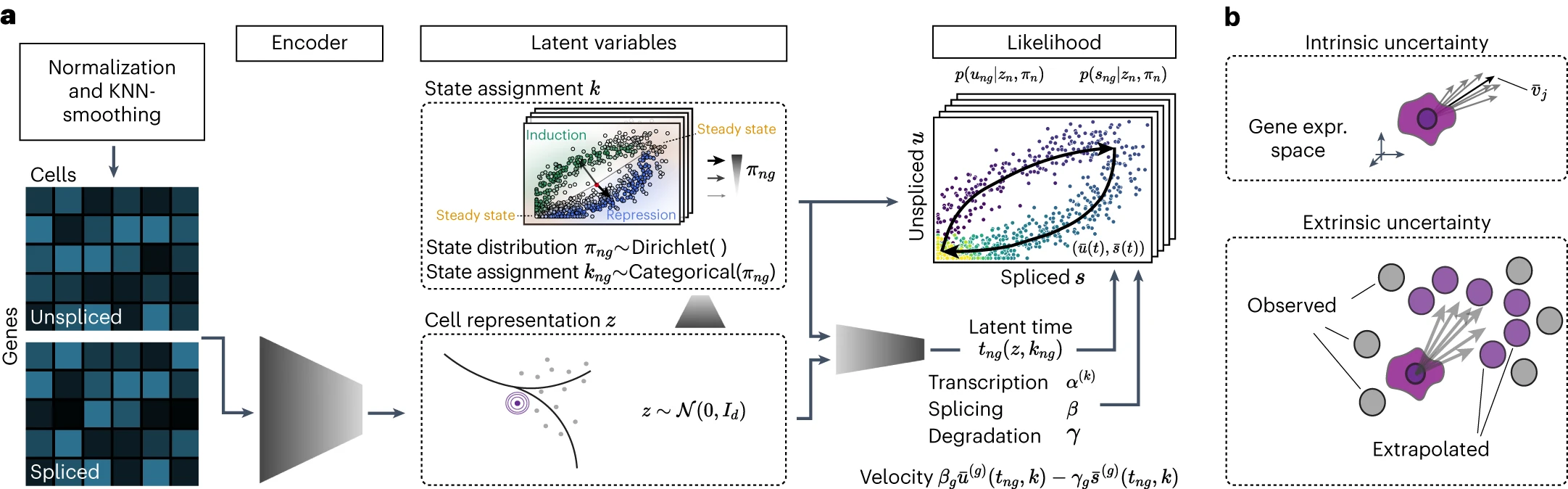
用于单细胞 RNA 速度分析的转录动力学深度生成模型
Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells. Nat Methods
- RNA 速度已被迅速用于指导快照单细胞数据中转录动力学的解释;然而,目前估计 RNA 速度的方法缺乏量化不确定性和确定对感兴趣系统的整体适用性的有效策略。
- 在这里,我们提出了 veloVI(速度变分推理),一种用于估计 RNA 速度的深度生成建模框架。 veloVI 学习 RNA 代谢的基因特异性动力学模型,并提供速度不确定性的转录组范围内的量化。
- 我们表明,在拟合优度、转录相似细胞之间的一致性以及用于量化 RNA 丰度的预处理管道的稳定性方面,veloVI 与之前的方法相比具有优势。此外,我们证明了 veloVI 的后验速度不确定性可用于评估速度分析是否适合给定的数据集。
- 最后,我们强调 veloVI 作为一个灵活的框架,通过调整底层动态模型以使用时间依赖性转录速率来建模转录动态。
类器官基因表达的时空、光遗传学控制
Spatiotemporal, optogenetic control of gene expression in organoids. Nat Methods
- 源自干细胞的类器官已成为研究人类发育和疾病建模的越来越重要的工具。然而,仍然需要控制和研究类器官中基因表达的时空模式的方法。
- 在这里,我们结合光遗传学和基因微扰技术,以可编程时空模式激活或敲低目标基因的 RNA。为了说明我们的方法的有用性,我们在人类神经发育的类器官模型中局部激活了 Sonic Hedgehog (SHH) 信号。
- 空间和单细胞转录组分析表明,这种局部诱导足以产生典型模式的类器官,并揭示了 SHH 对神经发育基因调控的贡献的新见解。
- 通过这项研究,我们提出光遗传学扰动与空间转录组学相结合,作为一种强大的技术来重新编程和研究类器官中的细胞命运和组织模式。
用脂质体抗生素杀死肿瘤相关细菌会产生诱导抗肿瘤免疫反应的新抗原
Killing tumor-associated bacteria with a liposomal antibiotic generates neoantigens that induce anti-tumor immune responses. Nat Biotechnol
- 越来越多的证据表明肿瘤微生物群是影响癌症进展的一个因素。在结直肠癌 (CRC) 患者中,我们发现针对厌氧菌的切除前抗生素可显着提高 25.5% 的无病生存率。对于小鼠研究,我们设计了一种封装在脂质体中的抗生素银-替硝唑复合物(LipoAgTNZ),以消除原发肿瘤和肝转移瘤中的肿瘤相关细菌,而不引起肠道微生物群失调。
- 被促肿瘤细菌(具核梭杆菌属)或益生菌(大肠杆菌尼氏菌属)定植的小鼠 CRC 模型对 LipoAgTNZ 治疗有反应,这使得两个感染具核梭杆菌的 CRC 模型的长期存活率超过 70%。抗生素治疗产生微生物新抗原,引发抗肿瘤 CD8+ T 细胞。异源和同源细菌表位有助于免疫原性,启动 T 细胞识别感染和未感染的肿瘤。
- 我们的策略针对肿瘤相关细菌来引发抗肿瘤免疫,为微生物组免疫治疗干预措施铺平道路。
雌激素受体-α 与 PNPLA3 p.I148M 变异之间的相互作用导致女性脂肪肝易感性
看上去似乎是一个挺简单的工作
- 由代谢功能障碍引起的脂肪肝病(FLD)是肝脏疾病的主要原因,并且患病率正在上升,尤其是在女性中。尽管在育龄期妇女可以免受 FLD 的侵害,但由于仍未知和未充分研究的原因,一些妇女在更年期会出现快速进展的疾病。
- 含 patatin 样磷脂酶结构域 3 (PNPLA3) p.I148M 变异占遗传性 FLD 变异的最大部分。在本研究中,我们发现女性与 PNPLA3 p.I148M 之间存在特定的乘性相互作用,以确定高危个体的 FLD(脂肪变性和纤维化,P < 10-10;晚期纤维化/肝细胞癌,P = 0.034) )和一般人群(丙氨酸转氨酶水平 P < 10-7)。在肥胖个体中,女性肝脏 PNPLA3 表达高于男性 (P = 0.007),并且小鼠中的表达与雌激素水平相关。
- 在人肝细胞和肝类器官中,PNPLA3 是由雌激素受体-α (ER-α) 激动剂诱导的。通过染色质免疫沉淀和荧光素酶测定,我们鉴定并表征了 PNPLA3 增强子内的 ER-α 结合位点,并通过 CRISPR-Cas9 基因组编辑证明该序列驱动 PNPLA3 p.I148M 上调,导致脂滴积累和纤维形成具有星状细胞的维度多谱系球体。
- 这些数据表明 ER-α 和 PNPLA3 p.I148M 变体之间的功能相互作用有助于女性 FLD。
共生酵母和食品来源酵母对交叉反应 T 细胞的选择驱动克罗恩病中细胞毒性 TH1 细胞反应
- CD4+ T 细胞针对肠道微生物的异常反应被认为是炎症性肠病中粘膜炎症的驱动因素。与疾病相关的微生物种类和相应的微生物特异性致病性 T 细胞表型仍然很大程度上未知。
- 在本研究中,我们确定了常见的肠道共生酵母和食物来源的酵母,它们是克罗恩病 (CD) 患者 CD4+ T 细胞反应改变的直接激活剂。 CD 中的酵母反应性 CD4+ T 细胞表现出细胞毒性 T 辅助细胞 (TH1 细胞) 表型,并显示 T 细胞克隆的选择性扩增,这些克隆与几种共生真菌以及食物来源的真菌物种高度交叉反应。这表明在慢性肠道疾病的情况下,通过反复遇到保守的真菌抗原而进行交叉反应性 T 细胞选择。
- 我们的研究结果强调了酵母作为 CD 患者异常 CD4+ T 细胞反应驱动因素的作用,并表明肠道驻留真菌共生体和酵母的日常饮食摄入可能有助于 CD 患者炎症性 CD4+ T 细胞反应的慢性激活。
D-LMBmap:用于神经回路全脑分析的全自动深度学习管道
D-LMBmap: a fully automated deep-learning pipeline for whole-brain profiling of neural circuitry. Nat Methods. full html
- 最近组织透明方法和光片荧光显微镜的扩散和集成为以极高的通量实现中尺度三维全脑连接图谱创造了新的机会。随着大型、高质量成像数据集的快速生成,下游分析正在成为中尺度连接组学的主要技术瓶颈。由于详尽的手动注释和高度定制的培训,当前的计算解决方案是劳动密集型的,应用有限。同时,全脑数据分析往往需要多个软件包的组合以及用户的二次开发。
- 为了应对这些挑战,我们开发了 D-LMBmap,这是一个端到端软件包,提供集成工作流程,包含三个基于深度学习算法的全脑连接映射模块:轴突分割、大脑区域分割和全脑配准。
- D-LMBmap 不需要对轴突分割进行手动注释,并在单个工作流程中实现全脑投影组的定量分析,并且在所有测试模式中对多种细胞类型具有卓越的准确性。
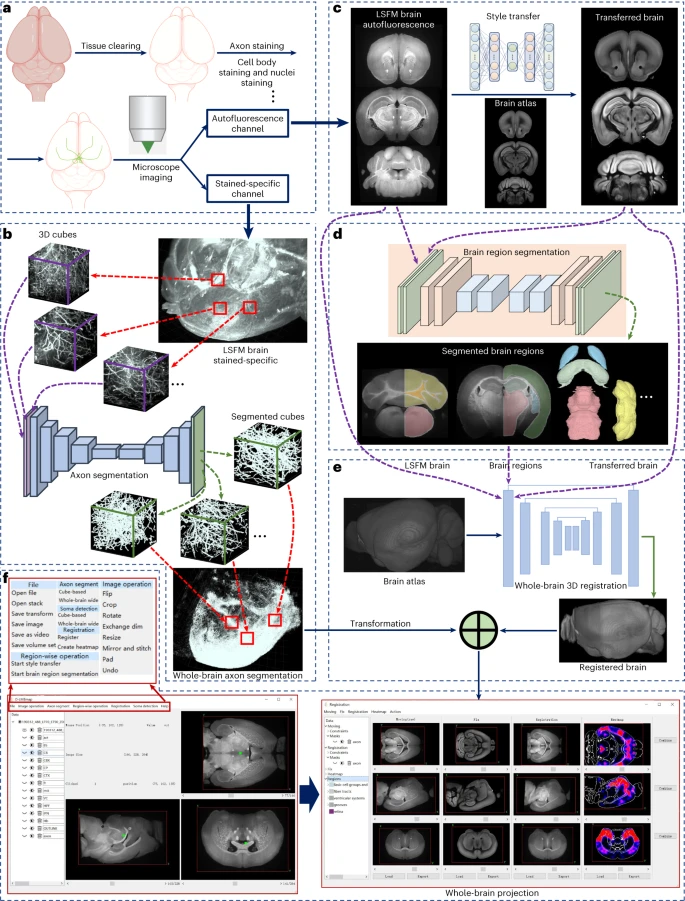
用于单分子定位显微镜的深度学习驱动的自适应光学器件
Deep learning-driven adaptive optics for single-molecule localization microscopy. Nat Methods
- 生物组织的不均匀折射率会模糊和扭曲单分子发射模式,从而产生图像伪影并降低单分子定位显微镜(SMLM)可实现的分辨率。传统的无传感器自适应光学方法依赖于迭代镜面变化和图像质量指标。然而,这些度量导致度量响应不一致,从而从根本上限制了它们在组织中像差校正的功效。
- 为了绕过迭代试验然后评估的过程,我们为 SMLM 开发了深度学习驱动的自适应光学器件,以允许直接推断波前畸变和近实时补偿。我们训练有素的深度神经网络监控单分子实验中的各个发射模式,推断它们共享的波前畸变,通过动态滤波器提供估计值,并驱动可变形镜来补偿样本引起的像差。
- 我们证明了我们的方法可以同时估计和补偿 28 个波前变形形状,并通过 >130 µm 厚的脑组织样本提高三维 SMLM 的分辨率和保真度。
酵母和人类细胞中简化且灵敏的单/双核糖体分析
Streamlined and sensitive mono- and di-ribosome profiling in yeast and human cells. Nat Methods
- 核糖体分析通过核糖体占用的转录组范围调查揭示了翻译的多种调控和扰动,并通过核糖体保护的信使 RNA 片段的测序读出。核糖体足迹的生成及其转化为测序文库在技术上要求很高,并且对扭曲生理核糖体占用表示的偏差敏感。
- 我们通过使用 P1 核酸酶而不是 RNase I 产生核糖体足迹,并用有序双模板中继取代 RNA 连接来解决这些挑战,这是一种用于测序文库制备的单管方案,通过逆转录结合接头。
- 我们的简化方法减少了序列偏差并增强了核糖体足迹相对于核糖体 RNA 的富集。此外,P1核酸酶保留了不同的并置核糖体复合物,这些复合物在翻译起始、停滞和终止过程中提供了有关酵母和人类核糖体命运的信息。
- 我们针对 mRNA 足迹生成和捕获的优化方法提供了更丰富的翻译组图谱,且输入量低且技术挑战更少。
机器学习/组学类
从纵向测序中识别神经胶质瘤进化的预测因子
Identifying predictors of glioma evolution from longitudinal sequencing. Sci Transl Med. full html
香港科技大学王吉光团队,主要方向之一就是肿瘤进化。
- 克隆进化驱动癌症进展和治疗耐药性。最近的研究揭示了神经胶质瘤的不同纵向轨迹,但指导治疗后癌症进化的早期分子特征仍不清楚。在这里,我们收集了 544 例成人弥漫性神经胶质瘤的初始-复发肿瘤对的测序和临床数据,并进行多变量分析,以确定三种弥漫性神经胶质瘤亚型中肿瘤进化的早期分子预测因子。
- 我们发现,初始诊断时 CDKN2A 缺失先于 IDH 突变神经胶质瘤后期发生的肿瘤坏死和微血管增殖。 IDH 野生型神经胶质瘤诊断时的 Ki67 表达与复发时获得的超突变呈正相关。在所有神经胶质瘤亚型中,诊断时的 MYC 增益或 MYC 靶点激活与复发时治疗诱导的超突变相关。
- 为了预测神经胶质瘤的进化,我们构建了 CELLO2(纵向数据癌症进化第 2 版),这是一种机器学习模型,集成了诊断时的特征,以预测治疗后的超突变和进展。 CELLO2 成功地将患者分为具有不同预后的亚组,并从低级别 IDH 突变非编码亚型中识别出一个以 MYC 增益为特征且进展后生存率较差的高风险患者组。然后,我们在神经胶质瘤细胞系和同基因患者来源的神经胶质瘤球中进行了慢性替莫唑胺诱导实验,并证明 MYC 通过促进超突变来驱动替莫唑胺耐药性。从机制上讲,我们证明,通过与开放染色质和转录活性基因组区域结合,c-MYC 增加了关键错配修复基因对治疗诱导突变的脆弱性,从而引发超突变。
- 这项研究揭示了治疗下癌症演变的早期预测因素,并为针对弥漫性神经胶质瘤的癌症动态的精准肿瘤学提供了资源。
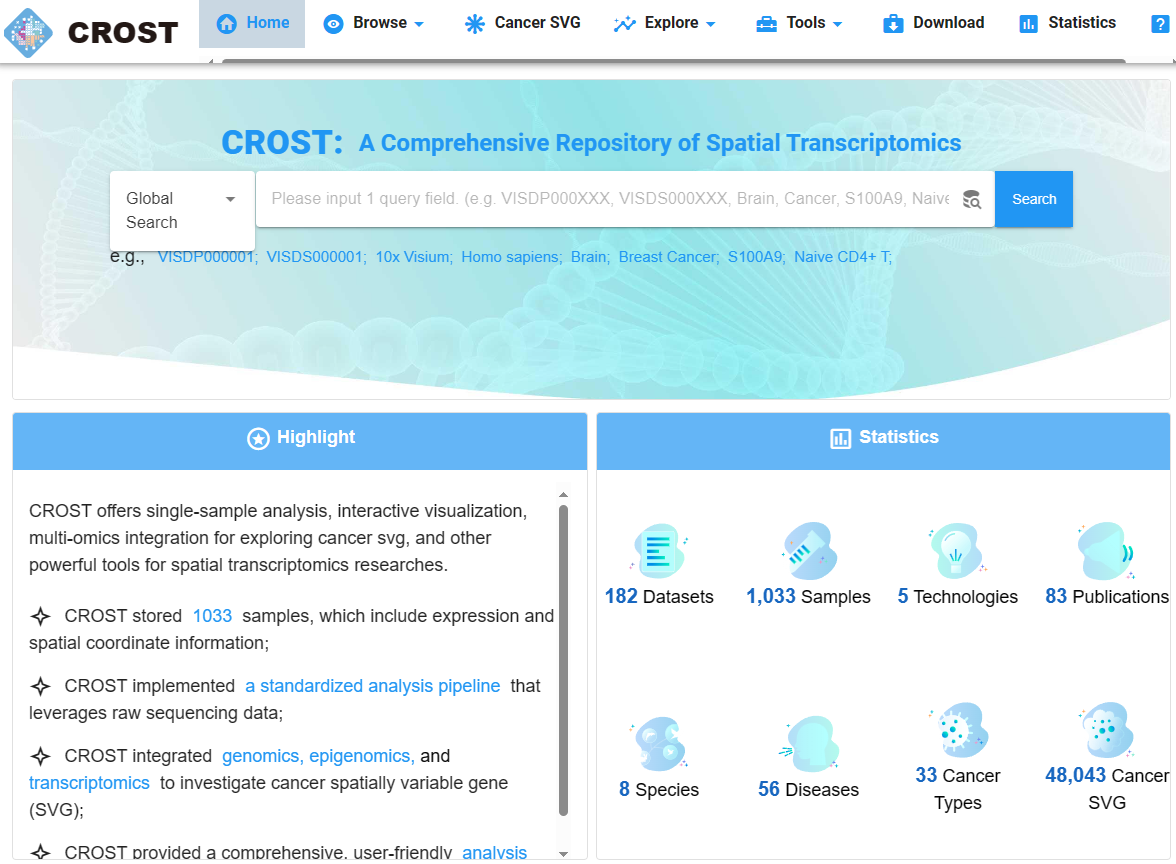
(~ ̄▽ ̄)~ CROST:空间转录组学的综合存储库
CROST: a comprehensive repository of spatial transcriptomics. Nucleic Acids Res
- 空间转录组测序技术的发展彻底改变了我们对复杂组织的理解,推动生命和健康科学进入空间组学时代。然而,当前用于访问和分析空间转录组数据的数据库的可用性是有限的。
- 为此,我们建立了空间转录组学综合存储库 CROST(https://ngdc.cncb.ac.cn/crost)。CROST 包含高质量样本,包含来自不同物种、器官和疾病的 182 个空间转录组数据集,其中包括 1033 个子数据集和 48043 个肿瘤相关空间可变基因 (SVG)。此外,它还包含标准化的空间转录组数据处理流程,集成单细胞 RNA 测序反卷积空间转录组数据,并评估相关性、共定位、细胞间通讯和生物功能注释分析。
- 此外,CROST 整合了转录组、表观基因组和基因组来探索肿瘤相关的 SVG,并全面了解它们在癌症进展和预后中的作用。此外,CROST还提供单样本基因集富集分析和SpatialAP两个在线工具,供用户对上传的空间转录组数据进行注释和分析。 CROST 的用户友好界面方便浏览、搜索、分析、可视化和下载所需信息。
- 总的来说,CROST 为组织结构提供了新鲜而全面的见解,并为理解疾病(特别是肿瘤组织)的多种生物学机制奠定了基础。
阿尔茨海默病 CSF 生物标志物谱的多变量 GWAS 表明 GRIN2D 在突触功能中的作用
- 背景:阿尔茨海默病 (AD) 的全基因组关联研究 (GWAS) 已确定了几个风险位点,但许多位点仍未知。脑脊液 (CSF) 生物标志物可能有助于基因发现,我们之前证明了六种 CSF 生物标志物(β-淀粉样蛋白、总/磷酸化 tau、NfL、YKL-40 和神经粒蛋白)聚类为五个主要成分 (PC),每个成分在统计上代表独立的生物过程。在这里,我们的目标是 (1) 识别与这些 CSF 谱相关的常见遗传变异,(2) 评估相关变异在 AD 病理生理学中的作用,以及 (3) 探索潜在的性别差异。
- 方法:我们在两项多中心研究(EMIF-AD 和 ADNI)中对五种生物标志物 PC 中的每一种进行了 GWAS。总共对 973 名参与者(n = 205 名对照者、n = 546 名轻度认知障碍患者、n = 222 名 AD)进行了 7,433,949 个常见 SNP 和 19,511 个蛋白质编码基因的分析。结构方程模型测试了生物标志物 PC 是否介导 AD 的遗传风险影响,分层和交互模型探讨了性别特异性影响。
- 结果:五个位点显示与 CSF 谱在全基因组范围内显着相关,其中两个是新的(rs145791381 [炎症] 和 GRIN2D [突触功能]),三个是先前描述的(APOE、TMEM106B 和 CHI3L1)。对独立数据集中两个新信号的后续分析仅支持 GRIN2D 基因座,该基因座包含几个功能上有趣的候选基因。中介测试表明,APOE 中的变异通过淀粉样蛋白和 tau 病理学相关过程与 AD 状态相关,而 TMEM106B 和 CHI3L1 中的标记仅通过神经元损伤/炎症与 AD 相关。此外,七个基因座显示出与 AD 生物标志物的性别特异性关联。
- 结论:这些结果表明通路和性别特异性分析可以提高我们对 AD 遗传学的理解,并可能有助于精准医学。
基于单细胞拷贝数改变破解肿瘤进化模式
Cracking the pattern of tumor evolution based on single-cell copy number alterations. Brief Bioinform
广东省心血管病研究所、广东省人民医院、广东省医学科学院医学研究所、广东省人民医院(广东省医学科学院)、南方医科大学。
- 拷贝数改变(CNA)是肿瘤发生和进展的关键特征。肿瘤发生过程中各种CNA的积累在驱动肿瘤进化中发挥着关键作用。由不同 CNA 驱动的异质克隆具有不同的选择优势,导致肿瘤进化的不同模式,这对于开发有效的癌症疗法至关重要。单细胞测序技术的最新进展使得能够以单细胞分辨率对肿瘤细胞群进行全基因组拷贝数分析。这使得探索CNA的进化模式并准确发现肿瘤内异质性的机制成为可能。
- 在这里,我们提出了一种两步统计方法,根据单细胞拷贝数概况区分肿瘤细胞群的中性、线性、分支和间断进化模式。我们使用各种模拟和真实的单细胞基因组和转录组数据集评估了我们的方法,证明了其在预测肿瘤进化模式方面的高精度和稳健性。
- 我们将我们的方法应用于 20 名乳腺癌患者的单细胞 DNA 测序数据,并观察到间断进化是乳腺癌的主要进化模式。当将该方法应用于从 132 名不同癌症患者获得的单细胞 RNA 测序数据时,得出了类似的结论。此外,我们发现差异性免疫细胞浸润与特定的进化模式相关。
- 我们研究的源代码可在 https://github.com/FangWang-SYSU/PTEM 获取。
scQTLbase:集成的人类单细胞 eQTL 数据库
scQTLbase: an integrated human single-cell eQTL database. Nucleic Acids Res
- 全基因组关联研究(GWAS)已经识别出许多与疾病和性状相关的遗传变异。然而,这些变体的功能解释仍然具有挑战性。表达数量性状位点 (eQTL) 已被广泛用于识别与疾病相关的突变,但它们只能解释 20-50% 的疾病相关变异。单细胞 eQTL (sc-eQTL) 研究提供了巨大的机会,可以通过扩展的 eQTL 规模和更高分辨率的转录调控来识别新的疾病风险基因。然而,目前还没有专门针对单细胞 eQTL 的综合数据库供用户搜索、分析和可视化。
- 因此,我们开发了 scQTLbase (http://bioinfo.szbl.ac.cn/scQTLbase),这是第一个集成的人类 sc-eQTL 门户,具有涵盖 57 种细胞类型和 95 种细胞状态的 304 个数据集。它包含约 1600 万个与细胞类型/状态基因表达显着相关的 SNP,以及来自 3333 种性状/疾病的约 69 万个与疾病相关的 sc-eQTL。此外,scQTLbase 还提供 sc-eQTL 搜索、UMAP 图中的基因表达可视化、基因组浏览器以及基于感兴趣的 GWAS 数据集的共定位可视化。 scQTLbase 为 sc-eQTL 提供了一站式门户,将显着推进疾病易感基因的发现。
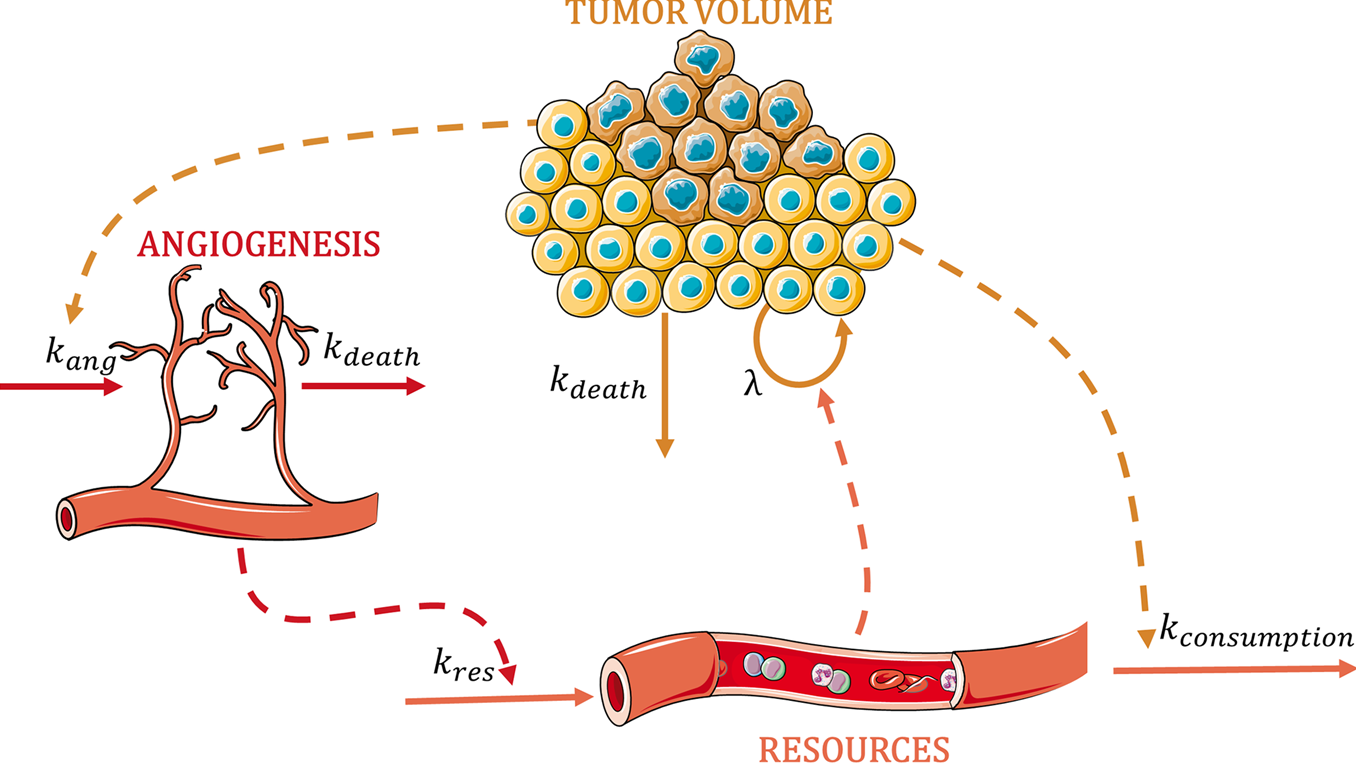
不受干扰的肿瘤生长动力学中振荡模式的机械表征:癌细胞与肿瘤微环境成分之间的相互作用
挺有趣的研究时间。实际上临床和生物学里比较少采用连续性模型来研究肿瘤生长,通常都是横断面研究(较长的时间间隔)。
- 临床前实验中未受干扰和受干扰的肿瘤生长动力学(TGD)的数学模型为建立转化框架提供了机会。最常用的不受干扰的肿瘤生长模型(即线性、指数、Gompertz 和 Simeoni)描述单调增长,尽管它们很好地捕捉了数据的平均趋势,但可以识别系统模型的错误指定。这代表了一个通过数学框架研究控制肿瘤生长动态的可能潜在机制的机会。
- 这项工作的总体目标是开发一种数据驱动的半机械模型,描述未经治疗的小鼠的非单调肿瘤生长。为此,将不同肿瘤类型和细胞系的纵向肿瘤体积分布汇集在一起并使用群体方法进行分析。在表征振荡模式(振荡器半周期在 8-11 天之间)并确认在不同的临床前实验中系统地观察到它们(p<10-9)后,建立了一个肿瘤生长模型,包括资源之间的相互作用(即氧或营养物质等资源之间的相互作用)、血管生成和癌细胞。
- 与之前使用的肿瘤生长模型相比,新结构除了改进了模型诊断(即 AIC 降低 71.48 且残差不存在自相关性 (p>0.05))之外,还允许以机械方式评估不同的肿瘤治疗方法方式。药物作用可能包含在肿瘤生长过程中发生的相关过程中。
- 简而言之,新模型除了描述肿瘤的非单调生长和肿瘤微环境的生物因素之间的相互作用外,还可用于探索临床前药物开发过程中单一疗法或联合疗法的不同药物场景。
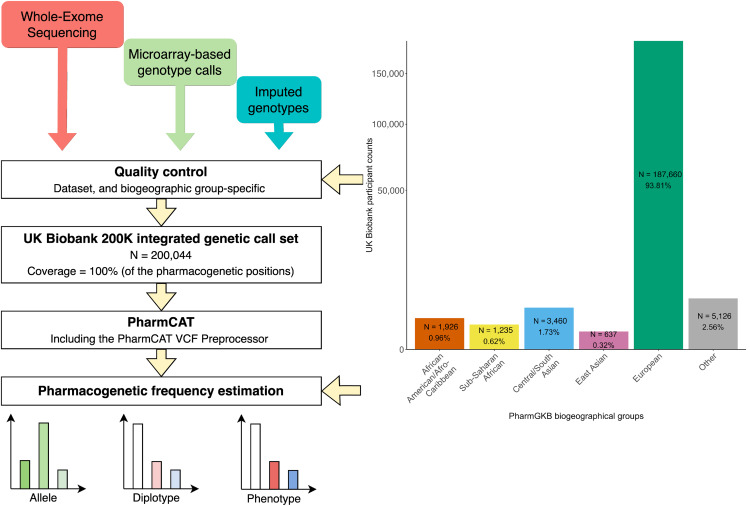
大型生物库中跨生物地理组的药物基因组等位基因的频率
Frequencies of pharmacogenomic alleles across biogeographic groups in a large-scale biobank – PubMed. Am J Hum Genet
- 药物基因组学(PGx)是精准医学的重要组成部分,有助于药物疗效的最大化和药物不良事件风险的降低。有关 PGx 等位基因频率的准确信息可改善 PGx 的实施。尽管如此,从已发表的等位基因数据中整理此类信息需要大量时间和资源。大多数研究中等位基因变异数量有限,导致某些等位基因被低估。
- 我们在集成的 20 万个英国生物库遗传数据集 (N = 200,044) 上应用了药物基因组学临床注释工具 (PharmCAT)。根据 PharmCAT 结果,我们估计了 5 个生物地理组中 17 个药物基因的 PGx 频率(等位基因、双倍型、表型和活性评分):欧洲、中/南亚、东亚、非洲-加勒比和撒哈拉以南非洲。每个生物地理组的 PGx 频率是不同的。即使具有相似表型比例的生物地理群体也是由不同组的显性 PGx 等位基因驱动的。 PharmCAT 还发现了以前研究中在某些群体中罕见或很少测试的“无功能”等位基因,例如加勒比非洲裔 (3.0%) 和撒哈拉以南非洲 (3.9%) 群体中的 SLCO1B1*31。估计的 PGx 频率通过 PharmGKB(药物基因组学知识库:www.pharmgkb.org)发布。
- 我们证明,英国生物银行等遗传生物银行是估计 PGx 频率的强大资源。提高我们对 PGx 等位基因和表型频率的理解,为未来的 PGx 研究和临床基因测试组设计提供指导,并更好地为来自更广泛生物地理背景的个体提供服务。
全基因组测序检测癌症易感性变异的临床效用和成本——多中心前瞻性队列研究
- 许多家庭和个人不符合已知遗传性癌症综合征的标准,但表现出不寻常的癌症群。这些家族可能携带癌症易感基因的致病变异,并且患癌症的风险较高。
- 这项多中心前瞻性研究招募了 195 名疑似患有遗传性癌症综合征的癌症患者,之前的临床靶向基因检测要么无法提供信息,要么无法进行。为了确定解释参与者表现的致病变异,进行了种系全基因组测序(WGS)和全面的癌症虚拟基因组分析。
- 在 5.1% (10/195) 的参与者中发现了与所呈现的癌症一致的致病变异,在另外 9.7% (19/195) 的参与者中发现了被认为具有潜在风险管理意义的次要发现的致病变异。健康经济分析估计,与随后进行 WGS 的标准测试(24,894 澳元)相比,采用虚拟面板的前期 WGS(8744 澳元)的每个病例的边际成本显着降低。财务分析表明,与传统检测相比,全国采用诊断性 WGS 检测将需要政府年度支出增加九倍。
- 这些发现证明用全基因组测序(WGS)取代传统检测可为癌症患者及其家庭带来临床上重要的益处。这种方法的采用将取决于不同付款人对负担能力的看法。
肝细胞癌TME的空间转录组学分析
经典研究套路
- 背景:新型免疫疗法联合疗法改善了肝细胞癌(HCC)患者的预后,但反应仅限于一小部分患者。对于 HCC 肿瘤微环境 (TME) 内细胞信号网络的肿瘤间和肿瘤内异质性知之甚少,而这是对现代全身治疗的反应的基础。
- 方法:我们应用空间转录组学 (ST) 分析来表征 HCC 切除标本中的肿瘤微环境,这些标本来自新辅助卡博替尼(一种主要阻断 VEGF 的多酪氨酸激酶抑制剂)和纳武单抗(一种 PD-1 抑制剂),15 名中的5名患者在切除时发现有病理反应。
- 结果:ST 分析表明,相对于无反应者,有反应肿瘤的 TME 富含具有促炎信号传导的免疫细胞和癌症相关成纤维细胞 (CAF)。反应肿瘤中丰富的癌症-免疫相互作用的特征是PAX5模块的激活,PAX5模块是B细胞成熟的已知调节因子,它与B细胞标记物表达增加的斑点共定位,表明这些细胞具有很强的活性。 HCC-CAF 相互作用在有反应的肿瘤中也丰富,并且与细胞外基质 (ECM) 重塑相关,因为肿瘤附近的 CAF 中 FOS 和 JUN 高度激活。 ECM 重塑与增殖性纤维化和免疫介导的肿瘤消退相关。在出现主要病理反应的患者中,有一名患者经历了早期 HCC 复发。该临床异常值的 ST 分析显示出明显的肿瘤异质性,具有独特的免疫不良肿瘤区域,类似于患者中无反应的 TME,其特征是 HCC-CAF 相互作用和癌症干细胞标记物的表达,可能介导早期肿瘤免疫逃逸以及该患者的复发情况。
- 结论:这些数据表明,HCC 对现代全身治疗的反应与独特的分子和细胞景观相关,并为增强和延长 HCC 全身治疗的反应提供了新的靶标。
空间转录组学揭示刺鼠再生耳朵对损伤的不对称细胞反应
- 与其他哺乳动物相比,刺鼠(Acomys)能够以无疤痕的方式再生皮肤和耳朵组织,包括毛囊、腺体和软骨。耳冲再生是不对称的,只有近端伤口侧参与再生。
- 在这里,我们表明,正常再生需要来自近端的线索,并使用空间分辨转录组学 (tomo-seq) 来了解该过程背后的分子和细胞事件。
- 通过分析整个耳朵的基因表达并比较近端和远端伤口侧之间的表达模块,我们识别了不对称基因表达模式并精确定位了空间和时间上的再生过程。此外,通过与非再生啮齿动物(小鼠、沙鼠)进行比较,我们强化了一个假设,即损伤引起的免疫反应的特殊性可能是多刺小鼠能够再生而其近亲却不能再生的关键决定因素之一。
- 我们的数据可在 SpinyMine 中获得,这是一种易于使用且可扩展的基于网络的工具,用于探索 Acomys 再生相关的基因表达。
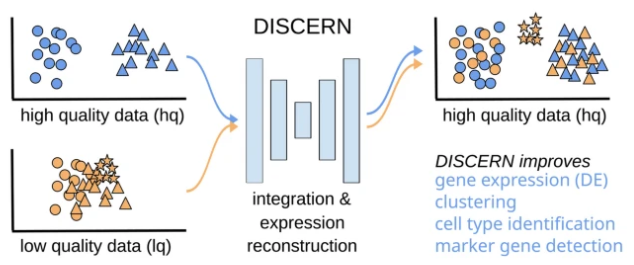
DISCERN:深度单细胞表达重建以改进细胞聚类以及细胞亚型和状态检测
DISCERN: deep single-cell expression reconstruction for improved cell clustering and cell subtype and state detection. Genome Biol
- 单细胞测序提供了对生物过程(包括细胞分化和身份)的详细了解。在提供深层细胞特异性信息的同时,该方法受到技术限制,最明显的是每个细胞的表达基因数量有限,这导致聚类和细胞类型识别不理想。
- 在这里,我们提出了 DISCERN,一种新颖的深度生成网络,它使用参考数据集精确重建缺失的单细胞基因表达。 DISCERN 在表达推断方面优于竞争算法,从而大大改善了细胞聚类、细胞类型和活性检测,并深入了解疾病的细胞调节。
- 我们证明 DISCERN 对于批次之间的差异具有鲁棒性,并且能够保持批次之间的生物学差异,这是插补和批次校正算法的常见问题。我们使用 DISCERN 检测两种看不见的 COVID-19 相关 T 细胞类型,即细胞毒性 CD4+ 和 CD8+ Tc2 T 辅助细胞,它们在不良疾病结果中具有潜在作用。我们利用患者血液的 T 细胞分数信息对轻度或重度 COVID-19 进行分类,AUROC 为 80%,可以作为疾病阶段的生物标志物。 DISCERN 可以轻松集成到现有的单细胞测序工作流程中。
- 因此,DISCERN 是一种使用参考数据集重建缺失的单细胞基因表达的灵活工具,并且可以轻松应用于各种数据集,从而产生新的见解,例如疾病机制。
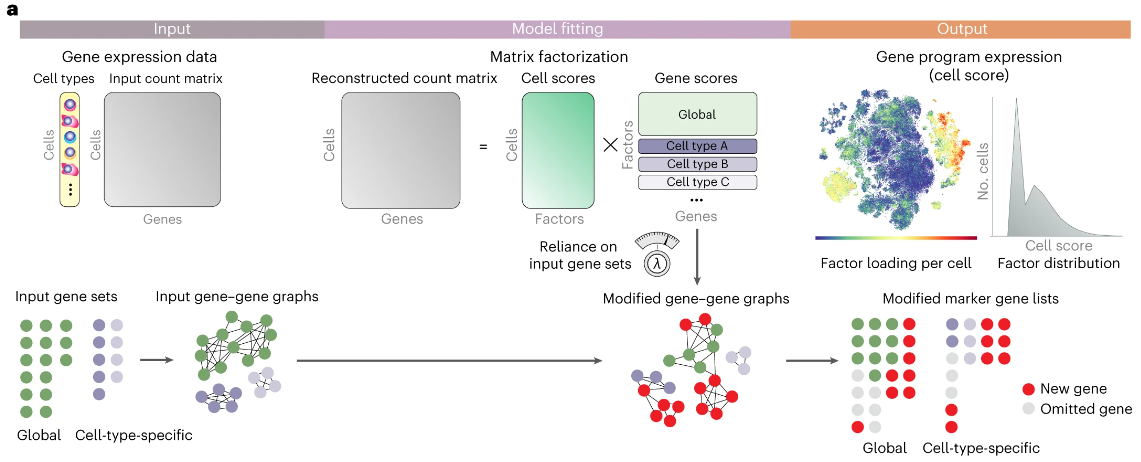
(~ ̄▽ ̄)~ 从单细胞数据中监督发现可解释的基因程序
Supervised discovery of interpretable gene programs from single-cell data. Nat Biotechnol
美国纪念斯隆凯特琳癌症中心-斯隆凯特琳研究所-计算和系统生物学项目
- 因子分析将单细胞基因表达数据分解为与样本中细胞执行的过程相对应的最小基因程序集。然而,矩阵分解方法容易出现技术问题且因子可解释性较差。
- 我们通过 Spectra 解决了这些问题,该算法将用户提供的基因程序与新程序的检测相结合,共同最好地解释表达共变。 Spectra 将现有的基因集和细胞类型标签合并为先验生物信息,显式地对细胞类型进行建模,并将输入基因集表示为基因-基因知识图,使用惩罚函数引导因子分解到输入图。
- 我们表明,Spectra 在具有挑战性的肿瘤免疫环境中优于现有方法,因为它发现了在免疫检查点治疗下发生变化的因素,解开了 CD8+ T 细胞肿瘤反应性和耗竭的高度相关特征,找到了一个程序来解释治疗下连续巨噬细胞状态变化,以及识别细胞类型特异性的免疫代谢程序。
使用 agoTRIBE 检测单细胞中转录组范围的 microRNA-靶标相互作用
Detection of transcriptome-wide microRNA-target interactions in single cells with agoTRIBE. Nat Biotechnol
- MicroRNA (miRNA) 根据其对目标转录本的选择,对众多生物过程发挥基因调控作用。目前可用于识别 miRNA 靶标的实验方法非常费力,并且需要数百万个细胞。
- 在这里,我们通过将 miRNA 效应蛋白 Argonaute2 与 ADAR2 的 RNA 编辑域融合,克服了这些限制,从而可以在单细胞中检测转录组范围内的 miRNA 靶标。 miRNA 引导融合蛋白到达其天然目标转录物,使它们经历 A>I 编辑,这可以通过敏感的单细胞 RNA 测序来检测。
- 我们证明 agoTRIBE 可以识别功能性 miRNA 目标,这得到了进化序列保守性的支持。在该方法的一项应用中,我们研究了单细胞中的 microRNA 相互作用,并确定了整个细胞周期中显着差异的靶向。 AgoTRIBE 还提供 RNA 丰度的转录组范围测量,并允许在单细胞水平上对复杂组织中的 miRNA 进行去卷积。
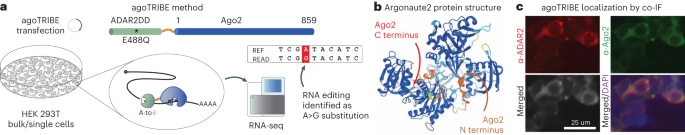
使用 geNomad 鉴定可移动遗传元件
Identification of mobile genetic elements with geNomad. Nat Biotechnol
- 了解测序数据的多样性、生态、生物技术应用以及对公共健康的影响是很重要的。
- 在这里,我们介绍 geNomad,一个分类和注释框架,它结合了基因内容和深度神经网络的信息来识别质粒和病毒的序列。 geNomad 使用超过 200,000 个标记蛋白谱的数据集来提供病毒基因组的功能基因注释和分类分配。使用条件随机场模型,geNomad 还可以高精度检测整合到宿主基因组中的原病毒。
- 在基准测试中,geNomad 对多种质粒和病毒实现了较高的分类性能(Matthews 相关系数分别为 77.8% 和 95.3%),大大优于其他工具。利用 geNomad 的速度和可扩展性,我们处理了超过 2.7 万亿个碱基对的测序数据,从而发现了可通过 IMG/VR 和 IMG/PR 数据库获得的数百万种病毒和质粒。
- geNomad 可在 https://portal.nersc.gov/genomad 上获取。
从全蛋白质组测量推断蛋白质复合物重塑的计算框架
A computational framework for the inference of protein complex remodeling from whole-proteome measurements. Nat Methods
系列研究
- 蛋白质复合物负责大多数细胞功能的制定。为了使蛋白质复合物形成并发挥作用,其亚基通常需要以规定的数量比例存在。通常,蛋白质复合物组成的整体变化是通过实验方法来评估的,这种方法往往非常耗时。
- 在这里,我们开发了一种计算算法 AlteredPQR,用于基于定量蛋白质组测量对亚基比率的系统评估来检测改变的蛋白质复合物。我们将其应用于乳腺癌细胞系和患者活检的测量,并能够在更具侵袭性的癌症中识别 HDAC2 表观遗传复合物的强烈重塑。
- 所提出的算法可作为 R 包使用,并通过从自下而上的蛋白质组数据集中提取功能相关信息来推断蛋白质复合物状态的变化。
在反向电泳力下线性化全长蛋白质通过工程纳米孔的易位
Translocation of linearized full-length proteins through an engineered nanopore under opposing electrophoretic force. Nat Biotechnol
荷兰-格罗宁根大学-格罗宁根生物分子科学与生物技术研究所
系列研究
- 纳米孔最近已被用于识别和指纹识别蛋白质。然而,由于蛋白质与 DNA 不同,不具有均匀的电荷,因此电泳力通常不能用于使它们移位或线性化。
- 在这里,我们表明,在 CytK 纳米孔的内腔中引入间隔约 1 nm 的电荷组会产生电渗流,从而诱导非结构化天然多肽在强电泳力的作用下进行单向运输。
- 分子动力学模拟表明,这种电渗主导的力在 -100 mV 时的强度约为 20 pN,这与单链 DNA 上的电力相似。未折叠的多肽在穿过纳米孔时会产生电流特征,可用于识别蛋白质。
- 这种方法可用于易位和拉伸蛋白质,以进行酶促和非酶促蛋白质鉴定和测序。
(~ ̄▽ ̄)~ 通过纳米孔明确区分所有 20 种蛋白氨基酸及其修饰
Unambiguous discrimination of all 20 proteinogenic amino acids and their modifications by nanopore. Nat Methods
这也是我听说三代测序后一直纳闷的一个问题。十分重要的论文。
系列研究
- 天然蛋白质由 20 种蛋白氨基酸及其翻译后修饰 (PTM) 组成。然而,由于缺乏合适的纳米孔传感器能够同时区分所有20种氨基酸及其PTM,利用纳米孔对蛋白质进行直接测序尚未实现。
- 在这里,我们提出了一种含有唯一 Ni2+ 修饰的工程异八聚耻垢分枝杆菌孔蛋白 A (MspA) 纳米孔。它能够完全区分所有 20 种蛋白氨基酸和 4 种代表性修饰氨基酸:Nω,N’ω-二甲基-精氨酸 (Me-R)、O-乙酰基-苏氨酸 (Ac-T)、N4-(β-N-乙酰基-D-葡萄糖胺基)-天冬酰胺(GlcNAc-N)和O-磷酸丝氨酸(P-S)。
- 在机器学习的辅助下,准确率达到了98.6%。还使用该策略分析了氨基酸补充剂片剂和肽酶消化的肽中的氨基酸。
- 这种同时区分所有 20 种蛋白氨基酸及其 PTM 的能力表明使用这种基于纳米孔的策略实现蛋白质测序的潜力。
通过汇集 CRISPR 筛选的可扩展单细胞 RNA 分析来剖析转录组动力学的关键调节因子
Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens. Nat Biotechnol
- 我们提出了一种组合索引方法 PerturbSci-Kinetics,用于在单细胞水平上捕获数百个遗传扰动的整个转录组、新生转录组和单向导 RNA (sgRNA) 身份。
- 我们对针对各种生物过程的汇集 CRISPR 筛选进行了分析,展示了 RNA 合成、加工和降解、miRNA 生物发生和线粒体 mRNA 加工过程中的基因表达调控,系统地解码了大规模 RNA 时间动态基础的全基因组调控网络。
使用 CellTag-multi 跨基因组模式的单细胞谱系捕获揭示了命运特异性基因调控变化
Single-cell lineage capture across genomic modalities with CellTag-multi reveals fate-specific gene regulatory changes. Nat Biotechnol
- 复杂的基因调控机制是分化和重编程的基础。现代单细胞谱系追踪 (scLT) 方法使用表达的、可遗传的 DNA 条形码将细胞谱系读数与单细胞转录组学结合起来。然而,对转录谱的依赖限制了对其他单细胞测定的适应。
- 借助 CellTag-multi,我们提出了一种方法,可以在单细胞 RNA 测序和使用测序分析进行转座酶可及染色质的单细胞分析中直接捕获表达为聚腺苷酸化转录本的可遗传随机条形码,从而可以对转录和转录进行独立克隆跟踪。
- 我们验证 CellTag-multi 来表征小鼠造血过程中祖细胞谱系启动的特征。此外,在成纤维细胞直接重编程为内胚层祖细胞的过程中,我们确定了靶向和脱靶命运的核心调控程序。此外,我们揭示了转录因子 Zfp281 作为重编程结果的调节因子,使细胞偏向脱靶间充质命运。我们的结果确立了 CellTag-multi 作为一种与多种单细胞模式兼容的谱系追踪方法,并证明了其在揭示不同分化和重编程范式中的命运特异性基因调控变化方面的实用性。
compleasm:更快、更准确地重新实现 BUSCO
compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics
- 评估基因完整性对于测量基因组组装的质量至关重要。不完整的组装可能会导致基因预测、注释和其他下游分析出现错误。 BUSCO 是一种广泛使用的工具,用于通过测试在各种分类单元中保守的一组单拷贝直向同源物的存在来评估基因组组装的完整性。然而,BUSCO 的速度很慢,尤其是对于大型基因组组装而言。将 BUSCO 应用于大量组件是很麻烦的。
- 在这里,我们提出了 compleasm,这是一种评估基因组组装完整性的有效工具。 Compleasm 利用 miniprot 蛋白质到基因组比对器和 BUSCO 的保守直系同源基因。它在人体组装方面比 BUSCO 快 14 倍,并且报告的完整性比 BUSCO 的 95.7% 更准确,为 99.6%,这与 T2T-CHM13 的注释完整性 99.5% 非常一致。
皮质中间神经元作为对抗性鉴别器的作用
A role for cortical interneurons as adversarial discriminators. PLoS Comput Biol
- 大脑从经验中学习感官信息的表征,但其执行此操作的算法仍然未知。一种流行的理论将表征形式化为感觉刺激生成模型中的推断因素,这意味着学习必须改进这种生成模型和推理过程。该框架是许多经典的感觉学习计算理论的基础,例如玻尔兹曼机、唤醒/睡眠算法,以及最近提出的大脑通过比较清醒和做梦活动的对抗性算法进行学习的建议。然而,为了让这些理论能够深入了解感觉学习的细胞机制,它们必须首先与大脑中介导它们的细胞类型联系起来。
- 在这项研究中,我们研究了皮质中间神经元的亚型是否可以通过充当鉴别器来介导感觉学习,鉴别器是表示学习的对抗性算法的关键组成部分。我们描述了这种中间神经元如何通过可塑性规则来表征,该可塑性规则从清醒状态下的赫布可塑性转变为梦境状态下的反赫布可塑性。评估该算法的计算优势和劣势,我们发现它擅长学习具有循环连接的网络中的表示,但随着网络规模的扩展却很差。如果网络也在诱发活动和生成样本之间以更快的时间尺度振荡,则可以部分解决此限制。
- 因此,我们提出以中间神经元作为鉴别器的对抗性算法是生物系统中感官学习的一种合理且可测试的策略。
评估大型语言模型解决生物信息学入门课程中的编程练习的能力
Evaluating a large language model’s ability to solve programming exercises from an introductory bioinformatics course. PLoS Comput Biol
研究了一个答案似乎显而易见的问题,这好吗?
- 计算机编程是生命科学家的基本工具,使他们能够执行重要的研究任务。然而,尽管做出了各种教育努力,学习编写代码对于生命科学学科的学生和研究人员来说仍然是一项具有挑战性的工作。人工智能的最新进展使得将人类语言提示翻译为功能代码成为可能,这引发了关于这些技术是否可以帮助(或取代)生命科学家编写代码的努力的问题。
- 使用生物信息学入门课程中的 184 个编程练习,我们评估了此类工具(OpenAI 的 ChatGPT)成功完成编程任务的程度。 ChatGPT 第一次尝试就解决了 139 题 (75.5%) 的练习。对于剩下的练习,我们向模型提供了自然语言反馈,促使它尝试不同的方法。在 7 次或更少的尝试内,ChatGPT 解决了 179 (97.3%) 的练习。
- 这些发现对生命科学教育和研究具有重要意义。教师可能需要调整他们的教学方法和评估技术,以适应公众可以获得的这些新能力。对于某些编程任务,研究人员也许能够与机器学习模型协作来生成功能代码。
生物分子节律分析方法CRICUST
- 昼夜节律系统驱动行为和生物过程近 24 小时的振荡。潜在的核心分子时钟调节其他基因的表达,研究表明,哺乳动物中超过 50% 的基因表达呈现 24 小时节律模式,特定基因的循环从一个组织到另一个组织都有所不同。确定作为单个时间点采样的人体组织中的节律基因表达模式面临一些挑战,包括重建高噪声数据的时间顺序。先前的方法已尝试在一个或少数组织中解决这些挑战,假设时钟基因进化保守性得以保留。
- 在这里,我们介绍 CIRCUST,一种用于分析分子节律的新颖的 CIRCular-robUST 方法,它依赖于循环统计,对噪声具有鲁棒性,并且比现有方法需要更少的假设。
- 接下来,我们针对四项已知采样时间的对照实验验证了该方法,最后,将 CIRCUST 应用于基因型组织表达 (GTEx) 数据集中的 34 个组织,旨在构建全面的人类每日节律基因表达图谱。
- CIRCUST 提供了一个灵活的框架来制定和解决与人体组织中分子节律分析相关的问题。
GTEx 样本的多组织 H3K27ac 分析将表观基因组变异与疾病联系起来
Multitissue H3K27ac profiling of GTEx samples links epigenomic variation to disease. Nat Genet
- 与复杂性状相关的遗传变异主要是非编码的,它们对基因调控活性的影响在很大程度上仍然未知。
- 为了解决这个问题,我们分析了来自基因型组织表达 (GTEx) 的 387 个大脑、心脏、肌肉和肺样本中组蛋白标记 H3K27ac 的表观基因组变异。我们用组织特异性活性模式注释了 282 k 个活性调节元件 (ARE)。我们鉴定了 2,436 个性别偏见的 ARE 和 5,397 个受遗传影响的 ARE,与跨组织的 130 k 遗传变异 (haQTL) 相关。我们整合遗传和表观基因组变异,通过揭示候选作用组织、驱动 SNP 和受影响的 ARE,为来自 55 个全基因组关联研究 (GWAS) 的疾病相关位点提供机制见解。最后,我们基于遗传学(gLink 分数)构建 ARE-基因链接分数,并展示其优先考虑 SNP-ARE-基因电路的独特能力。
- 总的来说,我们的表观基因组数据集、计算集成和机制预测为理解精神分裂症等人类疾病/特征的分子基础提供了宝贵的资源和重要的见解。
使用神经最佳传输学习单细胞扰动反应
Learning single-cell perturbation responses using neural optimal transport – PubMed. Nat Methods
挺有趣的,可以看看
- 理解和预测单细胞对化学、遗传或机械扰动的分子反应是生物学的核心问题。获得单细胞测量通常需要破坏细胞。这使得学习异质扰动响应具有挑战性,因为我们只观察扰动或未扰动细胞的不成对分布。
- 在这里,我们利用最优传输理论和最近出现的输入凸神经架构来提出 CellOT,这是一个通过映射这些不成对分布来学习单个细胞对给定扰动的响应的框架。根据 scRNA-seq 和多重蛋白质成像技术的分析,CellOT 在预测单细胞药物反应方面优于当前的方法。
- 此外,我们通过(1)预测暴露于干扰素-β 的狼疮患者和胶质母细胞瘤患者对帕比司他的 scRNA-seq 反应,说明 CellOT 在未见过的环境中具有良好的泛化能力; (2) 推断不同物种的脂多糖反应; (3) 模拟不同亚群的造血发育轨迹。
(~ ̄▽ ̄)~ 一种新的贝叶斯因子分析方法改进了单细胞 CRISPR 筛选中受扰动影响的基因和生物过程的检测
A new Bayesian factor analysis method improves detection of genes and biological processes affected by perturbations in single-cell CRISPR screening. Nat Methods. full html
算法似乎不难,有时间可以看看
- 成簇规则间隔短回文重复序列 (CRISPR) 筛选与单细胞 RNA 测序相结合已成为在单细胞水平上表征遗传扰动对整个转录组的影响的强大工具。然而,由于其稀疏性和复杂的结构,单细胞CRISPR筛选数据的分析具有挑战性。特别是,标准差异表达分析方法通常不足以检测受 CRISPR 扰动影响的基因。
- 我们为此类数据开发了一种统计方法,称为引导稀疏因子分析(GSFA)。 GSFA 推断代表共调控基因或基因模块的潜在因子;通过借用这些因素的信息,它推断出遗传扰动对个体基因的影响。
- 我们通过广泛的模拟研究证明,GSFA 检测扰动效应的能力比最先进的方法高得多。使用来自人类 CD8+ T 细胞和神经祖细胞的单细胞 CRISPR 数据,我们表明 GSFA 识别了生物学相关的基因模块和受 CRISPR 扰动影响的特定基因,其中许多基因被现有方法遗漏,从而为基因功能提供了新的见解参与 T 细胞激活和神经发育。
(~ ̄▽ ̄)~ 用于多组织基因表达插补的超图分解
Hypergraph factorization for multi-tissue gene expression imputation. Nat Mach Intell. full html
- 整合跨组织和细胞类型的基因表达对于理解驱动疾病和表征体内平衡的协调生物机制至关重要。然而,传统的多组织整合方法无法处理未收集的组织或依赖于基因型信息,而基因型信息通常不可用且存在隐私问题。
- 在这里,我们提出 HYFA(超图因子分解),一种参数高效的图表示学习方法,用于多组织和细胞类型基因表达的联合插补。 HYFA 与基因型无关,支持每个个体收集不同数量的组织,并施加强烈的归纳偏差以利用组织和基因的共享调控架构。
- 在GTEx项目数据的性能比较中,HYFA 实现了优于现有方法的性能,特别是当有多个参考组织可用时。 HYFA 估算的数据集可用于识别可复制的调控遗传变异 (eQTL),与原始不完整数据集相比具有显着优势。
- HYFA 可以加速组织和细胞型转录组生物存储库的有效且可扩展的整合。
显性在哺乳动物中很常见,与反式作用基因表达和选择性剪接有关
Dominance is common in mammals and is associated with trans-acting gene expression and alternative splicing. Genome Biol
- 显性和其他非加性遗传效应是由等位基因之间的相互作用产生的,历史上这些现象在数量遗传学中发挥着重要作用。然而,大多数全基因组关联研究(GWAS)假设等位基因具有相加作用。
- 我们系统地研究了三种哺乳动物种群(F2 杂交猪、大鼠异质种群和小鼠异质种群)中 574 个生理和基因表达性状的显性(此处代表任何非加性的位点内相互作用)和加性。优势占所有物种所有生理性状遗传变异的约四分之一。血液学和免疫学特征表现出最高的显性方差,可能反映了针对病原体的平衡选择。
- 尽管大多数数量性状位点 (QTL) 可检测为加性 QTL,但我们在猪、大鼠和小鼠中分别鉴定了 154、64 和 62 个新的显性 QTL,这些 QTL 无法检测为加性 QTL。类似地,尽管大多数顺式作用表达 QTL 是加性的,但基因表达表现出很大一部分显性方差,而反式作用 eQTL 则富集显性。导致显性生理 QTL 的基因不太可能与其 QTL 物理连接,而是通过反式作用显性 eQTL 发挥作用。此外,在异种大鼠中,数以千计的 eQTL 与具有复杂加性和显性结构的选择性剪接亚型相关,这表明可能存在显性机制。
- 虽然遗传力主要是加性的,但许多哺乳动物的遗传效应占主导地位,并且可能通过不同的机制产生。因此,在 GWAS 中考虑加性效应和显性效应对于提高功效和揭示因果关系是有利的。
大鼠大脑的 Waxholm 空间图集:支持数据分析和集成的 3D 图集
Waxholm Space atlas of the rat brain: a 3D atlas supporting data analysis and integration. Nat Methods
- 体积脑图谱越来越多地用于整合和分析从动物模型获得的各种实验神经科学数据,但直到最近,完整覆盖大鼠大脑的公开数字图谱一直缺失。
- 在这里,我们介绍了Waxholm Space大鼠大脑图谱的更新,这是一个全面的开放获取体积图谱资源。该大脑图谱注释了 222 个结构,其中 112 个是新的,57 个与之前的版本相比进行了修订。它提供了大脑皮层、海马区、纹状体苍白球区、中脑多巴胺能系统、丘脑细胞群、听觉系统和主要纤维束的详细地图。我们记录了注释背后的标准,并演示了如何使用具有相关工具和工作流程的图谱来支持实验大鼠大脑数据的解释、整合、分析和传播。
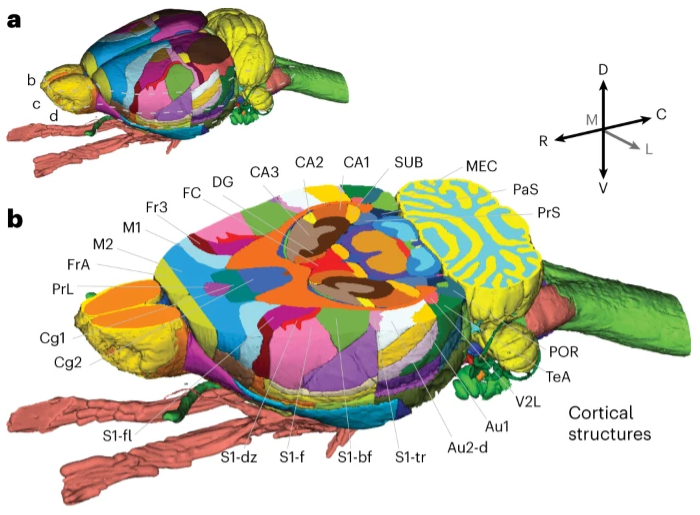
空间单细胞质谱法定义肝细胞蛋白质组的分区
Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome. Nat Methods
- 通过质谱法进行的单细胞蛋白质组学正在成为一种强大且公正的生物异质性表征方法。到目前为止,它仅限于培养细胞,而将该方法扩展到复杂组织将大大增强生物学洞察力。
- 在这里,我们描述单细胞深度视觉蛋白质组学 (single-cell Deep Visual Proteomics, scDVP),这是一种集成了高内涵成像、激光显微切割和多重质谱分析的技术。 scDVP 可解析细胞切片中当前深度为 1,700 个蛋白质的小鼠肝细胞的上下文相关空间蛋白质组。一半的蛋白质组以空间方式受到差异性调节,蛋白质水平在中央静脉附近发生显着变化。
- 我们将机器学习应用于蛋白质组类别和图像,随后仅从成像数据推断空间蛋白质组。 scDVP 适用于健康和患病组织,并补充其他空间蛋白质组学和空间组学技术。
使用计算机混合物对长读长 RNA 测序分析工具进行基准测试
Benchmarking long-read RNA-sequencing analysis tools using in silico mixtures. Nat Methods
- 由于缺乏具有内置事实依据的基准数据集,使得比较现有long-read同种型检测和差异表达分析工作流程的性能变得具有挑战性。
- 在这里,我们使用两种人肺腺癌细胞系进行了一项基准实验,每种细胞系均与合成的、剪接的、刺入的 RNA(亮片)一式三份进行分析。样品在 Illumina 短读长和 Oxford Nanopore Technologies 长读长平台上进行了深度测序。除了通过亮片获得的真实情况之外,我们还创建了计算机混合物样本,以便在没有真阳性或真阴性的情况下进行性能评估。
- 我们的结果表明,StringTie2 和 bambu 优于测试的六种异构体检测工具中的其他工具,DESeq2、edgeR 和 limma-voom 是测试的五种差异转录表达工具中最好的。对五种执行差异转录本分析的工具进行了比较,并没有明显的领先者,这表明该应用程序需要进一步开发方法。
(~ ̄▽ ̄)~ MBE:基于模型的差异测序数据富集估计和预测
MBE: model-based enrichment estimation and prediction for differential sequencing data. Genome Biol
加州大学伯克利分校电气工程与计算机科学系
有一定参考价值,要详细阅读
- 使用高通量测序数据来表征两种条件(例如有或没有药物暴露)之间的序列差异是一个普遍的问题,涉及量化序列丰度的变化,并预测未观察到的序列的此类差异。当前方法的一个主要缺点是它们在相关但不相同的read之间共享信息的能力极其有限。因此,它们无法有效地使用测序数据,也无法直接应用于许多感兴趣的环境中。
- 我们引入基于模型的富集分析(MBE)来克服这个缺点。我们使用模拟数据和真实数据来评估 MBE。总体而言,与当前的微分分析方法相比,MBE 提高了准确性。
GenArk:迈向百万个 UCSC 基因组浏览器
GenArk: towards a million UCSC genome browsers. Genome Biol
- 交互式图形基因组浏览器是基因组学中必不可少的工具,但它们并不包含所有最新的基因组组装。
- 我们从 NCBI 程序集创建 UCSC 基因组浏览器的基因组档案 (GenArk) 集合。建立在我们已建立的轨道中心系统的基础上,这可以实现注释的快速可视化。装配体带有基因模型、重复掩模、BLAT 和计算机 PCR。用户可以通过轨道中心和自定义轨道添加注释。我们可以批量导入第三方资源,通过 TOGA 和 Ensembl 基因模型演示了数百个程序集。
- https://hgdownload.soe.ucsc.edu/hubs/ 列出了三千二百六十九个 GenArk 程序集,并且可以在基因组浏览器网关页面上搜索。
(~ ̄▽ ̄)~ XA4C:通过自动编码器进行可解释的表示学习揭示关键基因
XA4C: eXplainable representation learning via Autoencoders revealing Critical genes. PLoS Comput Biol
有一定的参考价值。可以看看全文
- 机器学习模型经常用于转录组分析。特别是,表示学习(RL),例如自动编码器,可以有效地学习噪声数据中的关键表示。然而,学习到的表示,例如自动编码器中的“潜在变量”,很难解释,更不用说优先考虑功能后续的必需基因了。相比之下,在传统分析中,人们可以识别重要的基因,例如差异表达 (DiffEx)、差异共表达 (DiffCoEx) 和 Hub 基因。直观上,复杂的基因间相互作用可能超出了边际效应 (DiffEx) 或相关性(DiffCoEx 和 Hub)的捕获范围,这表明需要强大的 RL 模型。然而,缺乏可解释性和个体目标基因是强化学习在实践中广泛使用的障碍。
- 为了促进使用强化学习的可解释分析和基因识别,我们提出了“关键基因”,定义为对学习表示有很大贡献的基因(例如,自动编码器中的潜在变量)。
- 作为概念验证,在 eXplainable 人工智能 (XAI) 的支持下,我们实现了关键基因的 eXplainable 自动编码器 (XA4C),它可以量化每个基因对潜在变量的贡献,并据此对关键基因进行优先级排序。将 XA4C 应用到六种癌症的基因表达数据表明,关键基因捕获了癌症的重要途径。
- 值得注意的是,Critical 基因与 Hub 或 DiffEx 基因几乎没有重叠,但在综合疾病基因数据库(DisGeNET)和癌症特异性数据库(COSMIC)中具有更高的富集度,证明了其揭示大量未知生物学的潜力。例如,我们发现位于赖氨酸降解 (hsa00310) 途径中心的五个关键基因,在肿瘤和正常组织中显示出不同的相互作用模式。
- 总之,XA4C 有助于使用 RL 进行可解释的分析,而可解释的 RL 发现的关键基因可以促进复杂相互作用的研究。
造血基因治疗克隆追踪研究中细胞分化网络的可扩展推断
Scalable inference of cell differentiation networks in gene therapy clonal tracking studies of haematopoiesis. Bioinformatics
- 研究遗传性疾病下的细胞分化为改进当前的基因治疗策略提供了潜力。克隆追踪(Clonal tracking)为维持血细胞形成(称为造血过程)的群体干细胞动力学的数学建模提供了基础。然而,许多克隆跟踪协议依赖于细胞类型的子集来表征干细胞输出,并且生成的数据容易受到测量误差和噪声的影响。
- 我们提出了一个随机框架来从克隆跟踪数据推断细胞分化的动态模型。状态空间公式结合了描述细胞分化的随机准反应网络和考虑数据误差和噪声的高斯测量模型。我们开发了一种基于扩展卡尔曼滤波器、非线性优化和 Rauch-Tung-Striebel 平滑器的推理算法。
- 模拟表明,我们提出的方法优于最先进的方法,并且在节点大小和网络深度方面可以扩展到细胞分化的复杂结构。我们的方法在五项体内基因治疗研究中的应用揭示了细胞分化的不同动态。
- 我们的工具可以为生物学家和临床医生提供统计支持,以更好地了解基因治疗后的细胞分化和造血重建。可以修改状态空间模型的方程以推断除细胞分化之外的其他动力学。
- 随机框架在 R 包 Karen 中实现,可从 https://cran.r-project.org/package=Karen 下载。支持本研究结果的代码可在 https://github.com/delcore-luca/CellDifferentiationNetworks 上公开获取。
混杂泄漏:机器学习中混杂去除导致泄漏
Confound-leakage: confound removal in machine learning leads to leakage. Gigascience
有机会看看全文
- 机器学习 (ML) 方法是许多领域(包括流行病学和医学)现代数据分析的重要组成部分。非线性机器学习方法通常可以实现准确的预测,例如在个性化医疗中,因为它们能够对特征和目标之间的复杂关系进行建模。问题在于,机器学习模型及其预测可能会因特征中存在的混杂信息而产生偏差。为了消除这种虚假信号,研究人员经常采用特征线性混杂回归(CR)。虽然这被认为是处理混杂的标准方法,但在 ML 管道中使用 CR 可能存在的缺陷尚未完全理解。
- 我们提供的新证据表明,与一般预期相反,线性混杂回归与非线性 ML 方法结合使用时会增加混杂风险。使用一个使用目标作为混杂因素的简单框架,我们表明通过 CR 泄漏的信息可以增加零效应或中等效应,从而达到近乎完美的预测。通过对特征进行重组,我们提供了证据,证明这种增加确实是由于混杂泄漏而不是由于信息泄露。然后,我们在现实世界的临床应用中证明了混杂泄漏的危险,其中当使用抑郁症作为混杂时,使用语音衍生的特征来预测注意力缺陷/多动障碍的准确性被高估。
- 在构建机器学习模型时,由于混杂泄漏而处理不当甚至放大混杂效应,可能会导致不可信、有偏见和不公平的预测。我们揭露混杂泄漏陷阱并提供处理指南,可以帮助创建更强大、更值得信赖的机器学习模型。
ScSmOP:用于单细胞单分子多组学数据分析的通用计算管道
ScSmOP: a universal computational pipeline for single-cell single-molecule multiomics data analysis – PubMed. Brief Bioinform
- 单细胞多组学技术已广泛应用于检测细胞的关键特征。这些方法已经实现了单分子分辨率,甚至可以揭示空间定位。这些新兴方法提供了阐明个体细胞基因组、表观基因组和转录组异质性特征的见解。然而,它们给数据处理带来了新的计算挑战。
- 在这里,我们描述了单细胞单分子多组学管道(ScSmOP),这是一种用于条形码索引单细胞单分子多组学数据分析的通用管道。 ScSmOP本质上是利用C语言根据基于连接的条形码数据和基于合成的条形码数据建立基于间隔种子哈希表的条形码识别算法,然后进行数据映射和反卷积。我们在单细胞组学数据(scRNA-seq、scATAC-seq、scARC-seq)、单分子染色质相互作用数据(ChIA-Drop、SPRITE、RD- SPRITE)、单细胞单分子染色质相互作用数据(scSPRITE)以及来自各种细胞类型和物种的空间转录组数据展示了数据处理的高再现性。
- 此外,ScSmOP 显示出更快速的性能,是用于单细胞单分子多组学数据分析的多功能、高效、易于使用且强大的流程。
TransIntegrator:通过整合 Illumina 和 PacBio 转录组捕获几乎完整的蛋白质编码转录变体
TransIntegrator: capture nearly full protein-coding transcript variants via integrating Illumina and PacBio transcriptomes. Brief Bioinform
- 基因能够产生执行特定细胞功能的转录变体。然而,准确检测所有转录本变异仍然是一个长期存在的挑战,特别是在处理注释不良的基因组或没有已知基因组时。
- 为了解决这个问题,我们开发了一种新的计算方法 TransIntegrator,它能够在转录组范围内检测新的转录本变异。为此,我们确定了文昌鱼(一种具有重要进化意义的物种)连续胚胎发育阶段的 10 个 Illumina 测序转录组和一个 PacBio 全长转录组。基于转录组,我们利用 TransIntegrator 创建了一个综合的转录变异体库,即 iTranscriptome。由此产生的 iTrancriptome 包含 91,915 个不同的转录本变体,每个基因平均有 2.4 个变体。通过将基因数量从 21 954 个扩展至 38 777 个,显着改善了当前文昌鱼基因组注释。
- 进一步分析表明,基因扩展很大程度上归因于多个 Illumina 数据集的整合,而不是涉及 PacBio 数据。此外,我们还演示了 TransIntegrator 的一个示例应用,通过生成 iTrancriptome,帮助准确的转录组组装,其性能显着优于其他混合方法,例如 IDP-denovo 和 Trinity。
- 为了方便用户使用,我们将 TransIntegrator 的源代码存放在 GitHub 上,并在 Anaconda 中存放了 conda 包。总之,这项研究提出了一种经济实惠但有效的方法,用于大多数物种的可靠转录组研究。
(~ ̄▽ ̄)~ 机器学习用于个体化预测抗病毒药物根除丙型肝炎病毒后肝细胞癌的发展
思路挺简单的。预测使用的特征是什么?
- 背景和目的:在实现持续病毒应答(sustained viral response, SVR)后,对肝细胞癌(HCC)进行准确的风险分层对于最佳监测是必要的。我们的目标是开发和验证机器学习 (ML) 模型,以预测个体患者实现 SVR 后发生 HCC 的风险。
- 方法:在这项多中心队列研究中,纳入了 1742 名获得 SVR 的慢性丙型肝炎患者。开发了五种机器学习模型,包括 DeepSurv、梯度提升生存分析、随机生存森林 (RSF)、生存支持向量机和传统的 Cox 比例风险模型。使用 Harrel’ c 指数评估模型性能,并在独立队列(977 名患者)中进行外部验证。
- 结果:在 5.4 年的平均观察期内,122 名患者发展为 HCC(衍生队列中 83 名,外部验证队列中 39 名)。 RSF 模型在外部验证队列中使用七个参数实现 SVR(c 指数为 0.839)时显示出最佳辨别能力,并且当患者分为三个风险组时具有较高的辨别能力(P <0.001)。此外,该 RSF 模型可以通过在线应用程序为每位患者生成 HCC 发生的个性化预测曲线。
- 结论:我们开发并外部验证了 RSF 模型,该模型对于 SVR 后 HCC 风险具有良好的预测性能。这种新颖模型的应用可以在网站上找到。该模型可以提供数据来考虑有效的监测方法。需要进一步研究,为适合每个国家医疗状况的监测政策提出建议。
等位基因特异性 RNA N 6-甲基腺苷修饰揭示了人体组织中的功能性遗传变异
Allele-specific RNA N 6-methyladenosine modifications reveal functional genetic variants in human tissues. Genome Res
- 顺式和反式元件的复杂网络作用于 RNA N 6-甲基腺苷 (m6A),进而可能影响基因表达,并最终影响人类健康。要全面了解该网络,需要新的方法来准确测量由遗传变异引起的微妙 m6A 差异,其中许多变异与常见疾病有关。
- 为了解决这一差距,我们开发了一种方法,可以从 MeRIP-seq 数据中准确、灵敏地检测转录组范围内的等位基因特异性 m6A (ASm6A),并将其应用于发现 25 个人体组织中的 12,056 个高置信度 ASm6A 修饰。
- 我们还确定了 1184 个 ASm6A 调节的假定功能变体,我们通过实验验证了其中的一个子集。重要的是,我们发现许多与 ASm6A 相关的遗传变异在常见疾病相关和复杂性状相关风险位点上得到富集,并验证了两种疾病风险变异可以改变 m6A 修饰状态。
- 总之,这项工作提供了一种工具,可以在等位基因水平上理清 RNA 修饰的动态网络,并强调 m6A 和遗传学在人类健康和疾病中的相互作用。
转座子驱动后生动物锌指基因的进化
Transposable elements drive the evolution of metazoan zinc finger genes – PubMed. Genome Res
- Cys2-His2 锌指基因 (ZNF) 形成后生动物中最大的转录因子家族。 ZNF 进化是高度动态的,其特征是动物系统发育中众多亚科的快速扩张和收缩。 ZNF 快速进化背后的力量和机制仍然知之甚少,但越来越多的证据表明,在四足动物中,谱系特异性转座子 (TE) 的靶向和抑制在 Krüppel 相关盒 ZNF 的进化中发挥着关键作用。目前,尚不清楚这种功能和共同进化关系是否是 KZNF 所独有的,还是后生动物 ZNF 的更广泛特征。
- 在这里,我们提出的证据表明,基因组与 TE 的冲突是动物 ZNF 多样化的核心驱动因素。从 3221 个基因组组装中取样,我们发现逆转录元件的拷贝数与至少 7.5 亿年后生动物进化中 ZNF 的拷贝数相关。通过计算预测,我们发现 ZNF 在不同的动物物种中优先结合 TE。
- 我们进一步研究了在鲤鱼中发现的最大的 ZNF 亚家族,其特征是我们称为鱼 N 端锌指相关 (FiNZ) 结构域的保守序列。斑马鱼拥有大约 700 个 FiNZ-ZNF,其中许多在正选择下适应性进化。与哺乳动物 KZNF 一样,大多数斑马鱼 FiNZ-ZNF 在合子基因组激活开始时表达,并且在早期胚胎发生期间使用吗啉代阻断其翻译会导致转录活性 TE 的去抑制。
- 总之,这些数据表明 ZNF 多样化与整个动物进化过程中 TE 的扩展密切相关。
综述类
(~ ̄▽ ̄)~ 不断发展的癌症的治疗需要动态决策支持
Treatment of evolving cancers will require dynamic decision support. Ann Oncol. full pdf
美国H. Lee Moffitt 癌症中心及研究所-综合数学肿瘤学部、勒纳研究所-转化血液学和肿瘤学研究
- 癌症研究传统上侧重于开发新药物,但一个尚未充分探讨的问题是现有药物的剂量和频率。根据化疗早期建立的操作方式,大多数药物都是按照预定的时间表给药,旨在提供最大耐受剂量,并且仅根据毒性进行调整。然而,我们相信癌症的复杂性、不断演变的性质需要更加动态和个性化的方法。记录该领域的里程碑,我们表明,时间表选择的影响很大程度上取决于驱动治疗反应和失败的过程。因此,癌症的异质性和进化决定了不太可能出现一刀切的解决方案,相反,每个患者都应该制定最适合其当前疾病特征和治疗目标(即他们的“肿瘤景观”)的策略。
- 为了实现这种程度的个性化,我们需要数学建模。从这个角度来看,我们提出了一个五步“针对个性化肿瘤景观调整的自适应剂量(ADAPT)”范式,以整合跨尺度的数据和理解,并得出动态和个性化的时间表。最后,我们提供了模型引导的日程个性化的有希望的示例,并呼吁采取行动解决围绕数据收集、模型开发和集成的关键突出挑战。
开发个性化新抗原癌症疫苗的挑战
Challenges in developing personalized neoantigen cancer vaccines. Nat Rev Immunol
- 癌症免疫疗法最近的成功凸显了利用免疫系统治疗癌症的好处。疫苗在促进病原体免疫力方面有着悠久的历史,因此,针对癌症新抗原的疫苗一直被认为是指导和放大针对肿瘤的免疫反应,同时保护健康组织的工具。近年来,广泛的临床前研究和一百多项临床试验测试了新抗原发现和疫苗配方的不同策略。
- 然而,尽管人们对新抗原疫苗充满热情,但大多数临床试验仍然无法证明明确的功效。在这篇综述中,我们重点关注与疫苗设计和肿瘤环境有关的关键障碍,这些障碍仍有待克服,以释放新抗原疫苗在癌症治疗中的真正潜力。
癌症的动力学特征:黑色素瘤的表型转换和上皮间质可塑性
Dynamical hallmarks of cancer: phenotypic switching in melanoma and epithelial-mesenchymal plasticity. Semin Cancer Biol
- 表型可塑性最近被纳入癌症的标志。这种可塑性可以沿着许多相互关联的轴表现出来,例如干性和分化、药物敏感和耐药状态以及上皮细胞和间充质细胞状态之间。尽管人们越来越多地接受表型可塑性作为癌症的标志,但这一过程的动力学仍然知之甚少。特别是,预测性理解单个癌细胞和细胞群如何响应当前和过去环境刺激的强度和/或持续时间动态转换其表型所需的知识仍然远未完成。在这里,我们使用两个例子从系统水平的角度介绍了表型可塑性的最新研究:癌症中的上皮间质可塑性和黑色素瘤中的表型转换。
- 我们强调综合计算实验方法如何帮助揭示不同癌症表型可塑性的特定动态标志,以解决以下问题:a)存在多少种不同的细胞状态或表型? b)这些细胞状态之间的转变的可逆性如何,以及哪些因素控制可逆性的程度? c) 细胞间通讯如何能够改变细胞状态转换的速率并实现不同的表型异质性模式?了解表型可塑性的这些动态特征可能是将癌症治疗范式从反应性转变为更具预测性、主动性的方法的关键组成部分。
结直肠癌的微生物景观
The microbial landscape of colorectal cancer. Nat Rev Microbiol
- 结直肠癌(CRC)是全球发病率和死亡率的重要来源,迫切需要改进预防和治疗策略。随着我们对结直肠癌了解的加深,越来越明显的是,肠道微生物群由数万亿与结肠直接接触的微生物组成,在结直肠癌的发生和进展中发挥着重要作用。了解个体微生物和复杂微生物群落在结直肠癌发病机制中所起的作用及其伴随机制,将有助于产生新的结直肠癌预防和治疗干预措施。
- 在这篇综述中,我们讨论了有关 CRC 肠道微生物群整体扰动的最新证据、特定微生物与 CRC 的关联、微生物潜在驱动 CRC 发展的潜在机制以及复杂微生物群落在 CRC 发病机制中的作用。
- 尽管近年来我们对微生物群与结直肠癌之间关系的了解有所提高,但我们的研究结果凸显了当前研究中存在的巨大空白,需要填补这些空白才能将这些知识用于造福患者。
通过内窥镜筛查降低结直肠癌发病率
Reduction in colorectal cancer incidence by screening endoscopy. Nat Rev Gastroenterol Hepatol
德国癌症研究中心 (DKFZ) 临床流行病学和衰老研究部
- 在结肠镜检查广泛普及的时代,美国老年人群的结直肠癌 (CRC) 发病率下降了 50%,尽管 CRC 危险因素呈不利趋势,而且年轻时的 CRC 发病率有所增加。然而,NordICC 研究的随机试验报告的第一个结果表明,筛查结肠镜检查的效果相当有限。
- 正如本视角所述,现实世界和试验证据之间的明显差异可以解释为筛查内窥镜检查试验的效果估计值因多种因素而大幅减弱,包括有限的筛查依从性、在筛查范围之外广泛采用结肠镜检查以及纳入报告的事件病例数中普遍存在的、不可预防的结直肠癌病例。
- 考虑到患病率偏差的筛查内窥镜试验结果的其他解释与提供 CRC 筛查的国家 CRC 发病率降低的趋势一致,并且应鼓励更广泛地实施和采用有效的 CRC 筛查。
体育锻炼对抗癌免疫力的影响
The effect of physical exercise on anticancer immunity. Nat Rev Immunol
- 定期的体育活动与较低的癌症发病率和死亡率以及较低的肿瘤复发率相关。流行病学证据得到动物模型临床前研究的支持,表明定期锻炼可以延缓癌症的进展,包括高度侵袭性恶性肿瘤。
- 尽管运动抗肿瘤作用的机制仍有待确定,但癌症免疫监视的改善可能很重要,因为运动会刺激不同的免疫细胞亚型渗透肿瘤。还有证据表明,运动后收集的血液中的免疫细胞可以用作癌症的过继细胞疗法。
- 在本视角中,我们讨论了肌肉活动对于维持健康的免疫系统的重要性,并讨论了单次运动(即“急性”运动)和“常规”运动(即重复运动)的效果抗癌免疫,包括肿瘤浸润。我们还讨论了这一新兴研究领域的假设机制和临床意义。
使用工程化和内源效应器进行精确 RNA 碱基编辑
Precision RNA base editing with engineered and endogenous effectors. Nat Biotechnol
- RNA碱基编辑是指在完整RNA分子内重写遗传信息,具有多种功能,例如逃避内源性免疫系统和调节蛋白质功能。为了实现这一目标,在人类细胞中发现了某些酶,可以催化一种核碱基转化为另一种核碱基。这种自然过程可用于操纵和重新编码目标转录本中的任何碱基。与 DNA 碱基编辑相反,RNA 中引入的类似变化不是永久性的或可遗传的,而是允许可逆和可剂量的效应,从而吸引各种治疗应用。目前RNA碱基编辑的实践涉及腺苷和胞苷的脱氨基,分别转化为肌苷和尿苷。
- 在这篇综述中,我们总结了当前的定点 RNA 碱基编辑策略,并重点介绍了在提高编辑效率、精度、密码子靶向范围和体内递送至疾病相关组织方面取得的最新成就。除了工程编辑效应器之外,我们还重点关注利用作用于 RNA (ADAR) 酶的内源性腺苷脱氨酶的策略,并讨论将这些工具应用于基础研究和作为治疗方式的局限性和未来前景。我们预计该领域很快就会实现第一种 RNA 碱基编辑药物,很可能针对一种明确的遗传疾病。然而,长期的挑战将是找到该技术的最佳点,利用其独特的能力以安全和可剂量的方式调节信号线索、新陈代谢或其他临床相关过程。
(~ ̄▽ ̄)~ 液体活检联盟
The Liquid Biopsy Consortium: Challenges and opportunities for early cancer detection and monitoring. Cell Rep Med
- 液体活检这一新兴领域处于癌症和其他疾病新型诊断策略的前沿。液体活检可以对癌症进行微创分子表征,以进行诊断、患者分层治疗和纵向监测。液体活检策略包括检测和监测循环肿瘤细胞、游离 DNA 和细胞外囊泡。
- 在这篇综述中,我们探讨了现有基于液体活检的模式在癌症诊断和监测中的当前理解和作用。我们特别关注与液体活检和生物标志物开发相关的技术和临床挑战,该挑战由美国国家癌症研究所建立的液体活检联盟解决。
- 液体活检联盟开发了新的方法/测定法,并验证了现有的方法/技术来捕获和表征肿瘤衍生的循环货物,并解决了现有的挑战,并为推进生物标志物测定法提供了建议。
扩张型心肌病:原因、机制以及当前和未来的治疗方法
Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet
- 扩张型心肌病通常被定义为在不存在异常负荷条件(例如原发性瓣膜疾病)或足以引起心室重塑的显着冠状动脉疾病的情况下存在左心室或双心室扩张或收缩功能障碍。这一定义被认为过于严格,因为不伴有扩张的左心室运动功能减退可能是扩张型心肌病的最初表现。
- 扩张型心肌病的病因包括遗传因素(原发性扩张型心肌病)或后天因素(继发性扩张型心肌病)。获得性因素包括感染、毒素、癌症治疗、内分泌疾病、妊娠、快速心律失常和免疫介导的疾病。 5-15% 的获得性扩张型心肌病患者携带可能的致病或致病基因变异(即基因突变)。因此,诊断测试和治疗方法应始终考虑遗传因素和后天因素。
- 本次研讨会将重点关注当前的多维诊断和治疗方法,并讨论潜在的病理生理学,这些病理生理学可能推动未来的治疗,旨在修复或替换现有的基因突变,或针对遗传性或获得性扩张的特定炎症、代谢或促纤维化驱动因素心肌病。
构建智能 CAR T 细胞疗法:克服当前挑战的途径
Building smart CAR T cell therapies: The path to overcome current challenges. Cancer Cell
- 成功实施癌症过继细胞疗法(ACT)需要全面应对生物学和实际挑战。这种方法在很大程度上被忽视,导致 ACT 的潜力与其实际效果之间存在差距。
- 我们总结了创造“理想”ACT 产品最有前途的技术策略,重点关注嵌合抗原受体(CAR)工程细胞。由于有效 ACT 的许多要求对于大多数癌症来说都是常见的,因此我们在此概述的内容可能会产生更广泛的影响。
流行病学类
- 暂无。
---------------
完结,撒花!如果您点一下广告,可以养活苯苯😍😍😍
